La IA está cambiando el método científico. Durante siglos, la humanidad ha observado fenómenos naturales, ha inducido leyes y las ha comprobado posteriormente mediante evidencia experimental. Newton vio cómo caía una manzana, e imaginó que existía una fuerza que la atraía hacia el centro de la tierra. Postuló que esa fuerza debía depender de la masa de los cuerpos, e indujo una ley (la de la gravedad) que se demostró cierta en todas las medidas posteriores: todos los cuerpos caen, y todos lo hacen según esa ley, que plasmó en una simple ecuación matemática. Exactamente la misma ecuación explica la órbita de la luna o el movimiento de las galaxias. A partir de las teorías descubiertas, podemos hacer predicciones sobre los fenómenos naturales, sociales o económicos. Pero la IA puede prescindir de ese paso: observando series de datos, captura el patrón subyacente y realiza predicciones sin necesidad de explicitar las leyes que rigen los fenómenos. Cuando la IA crea un vídeo de cómo una piedra cae en la superficie de un lago, y genera las ondas de agua con total realismo, no utiliza fórmulas físicas para calcular el movimiento. Simplemente, proyecta una dinámica que ha aprendido por entrenamiento (viendo miles de vídeos previos), un “modelo del mundo”. La IA predice eventos complejos sin expresar las fórmulas que los rigen.
Gracias a eso, la industria farmacéutica acorta drásticamente los tiempos de desarrollo de fármacos. Hace un siglo, el descubrimiento de un antibiótico como la penicilina revolucionó la medicina. Hoy, la IA predice instantáneamente qué nuevos antibióticos serán efectivos para matar bacterias mutantes. AlphaFold, un sistema desarrollado por DeepMind (la división de IA de Google), predice la estructura tridimensional de las proteínas a partir de su secuencia genética, un problema central de la biología que llevaba más de 50 años sin resolverse. No conocemos el modelo subyacente que explica el fenómeno, pero la IA transforma un cuello de botella experimental (lento, caro y complejo) en un ejercicio computacional, acelerando de forma radical la investigación en medicina y el diseño de fármacos. AlphaFold ha puesto a disposición pública estructuras de cientos de millones de proteínas, muchas antes totalmente desconocidas. Si saber la forma de una proteína (cosa crítica para determinar su eficacia) requería años de I+D en laboratorio (el equivalente a un doctorado), hoy puede predecirse en minutos en un PC. Este cambio profundo en la forma de hacer química y biología molecular fue determinante para el Nobel de Química 2024, concedido a ingenieros computacionales de Google.
Pero también podemos pedir a la IA que nos ayude a descubrir las leyes ocultas que hay tras los fenómenos, realizando investigación fundamental. AlphaProof, otro sistema de DeepMind, ayuda a los matemáticos a descubrir algoritmos novedosos y soluciones a problemas sin resolver. Durante siglos, las matemáticas han avanzado gracias al trabajo paciente de miles de investigadores, y a unos pocos genios que dan saltos significativos (Gauss, Hilbert, Gödel…). Hoy, sistemas de IA alcanzan el nivel de medalla de oro en las Olimpiadas Internacionales de Matemáticas, la competición global más prestigiosa para jóvenes matemáticos, donde deben resolver problemas excepcionalmente complejos en álgebra, geometría o teoría de números. Sus soluciones son “sorprendentes, claras, precisas y, en su mayoría, simples”, según los evaluadores. La IA aborda la resolución de los problemas de Erdos, legendario matemático húngaro que listó más de 200 problemas complejos jamás resueltos. En pocos meses, 15 de ellos han pasado de “abiertos” a “resueltos”, la mayor parte con apoyo de la IA. Terence Tao, uno de los matemáticos más famosos del momento, considera la IA como una “auténtica extensión del razonamiento humano”.
Pensábamos que la naturaleza es bella porque es simple. Einstein descubrió una ley natural con su famosa ecuación E= mc2. La energía es igual a la masa por la velocidad de la luz (c) al cuadrado. Esa sencilla fórmula inaugura la era atómica: de un gramo de masa se puede obtener una cantidad ingente de energía. O, dicho de otro modo, la masa es energía hiperconcentrada. Una fórmula con solo tres variables, descubierta por uno de los grandes genios de la historia de la humanidad, cambió la trayectoria de la propia humanidad. ¿Es la naturaleza tan bella y armónica que las principales leyes naturales se expresan con fórmulas muy simples? ¿O quizá la capacidad cognitiva de los humanos, incluso de los más inteligentes, es tan limitada que únicamente (hasta ahora) hemos descubierto las leyes más simples? A la búsqueda de nuevas fórmulas complejas se ha lanzado la administración Trump con su proyecto Génesis, una ambiciosa misión que se ha comparado al proyecto Manhattan (que desarrolló la bomba atómica). Génesis pretende construir una gran plataforma de supercomputación e IA, alimentada con volúmenes masivos de datos de observaciones científicas, para acelerar el descubrimiento de nuevas leyes físicas, potenciar la innovación en ámbitos críticos y liderar la I+D global. La ciencia aumentada por IA adquiere así una dimensión geoestratégica.
¿Se está agotando el modelo clásico de la ciencia? La mayor parte de los científicos del planeta van a utilizar IAs de forma intensiva. Vamos a ver una explosión de productividad científica. Se anticipa una época de grandes descubrimientos. Realizar una investigación, comprobar una hipótesis o descubrir una nueva teoría será cada vez más fácil y más rápido gracias a la superinteligencia digital. Aunque, paradójicamente, la carrera científica puede colapsar. Las editoriales están recibiendo una avalancha de nuevos artículos científicos, asistidos por IA, que no tendrán capacidad de evaluar si no es mediante otra IA. ¿”Papers” de investigación escritos por IA y revisados por IA? ¿Vamos hacia una investigación autónoma y sin intervención humana? ¿Una ciencia sin científicos? La investigación del siglo XXI puede ser la más productiva de la historia, pero desarrollar una carrera científica será cada vez más selectivo y complicado. Nos esperan tiempos tan fascinantes como inquietantes
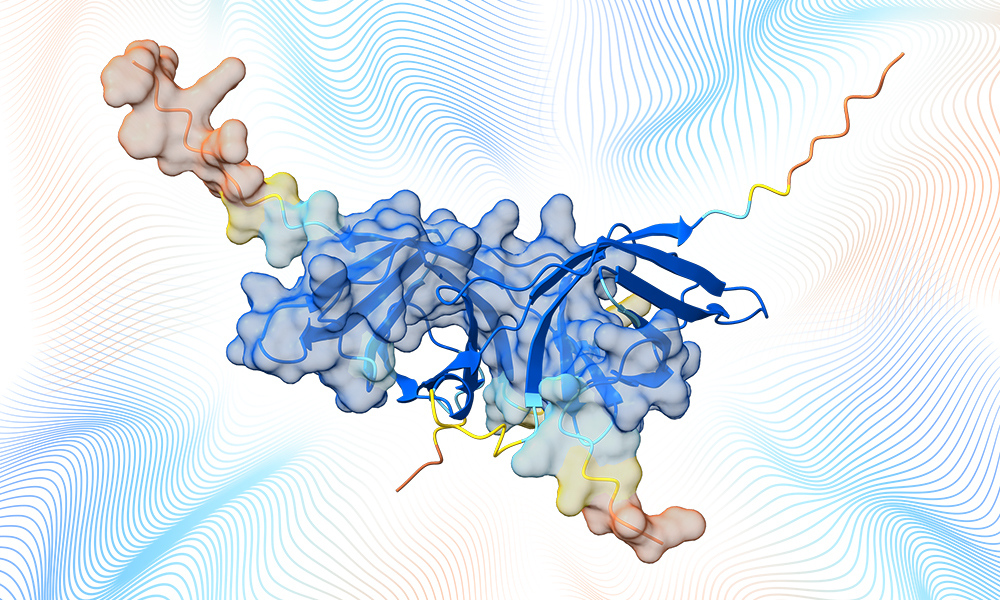
Hoy, nos complace compartir una actualización sobre nuestro avance hacia una nueva frontera en el diseño de fármacos. Hemos desbloqueado un nuevo paradigma de precisión predictiva para entender nuestro mundo biomolecular, lo que nos permite diseñar racionalmente nuevos medicamentos en un ordenador con una comprensión y precisión sin precedentes.
Estamos dando un vistazo a un subconjunto de las potentes y expansivas capacidades del Isomorphic Labs Drug Design Engine (IsoDDE), un sistema unificado de diseño computacional de fármacos, que avanza más allá de AlphaFold 3 (AF3) en su precisión predictiva e introduce nuevas capacidades que sirven de puente entre la predicción de estructuras y el descubrimiento real de fármacos.
Demostramos que nuestro IsoDDE más que duplica la precisión de AlphaFold 3 en un desafiante benchmark de generalización de la estructura proteína-ligando, predice afinidades de unión de pequeñas moléculas con precisiones que superan los métodos de referencia en física a una fracción del tiempo y coste, y es capaz de identificar con precisión nuevos bolsillos de unión en proteínas objetivo utilizando únicamente la secuencia de aminoácidos como entrada.
IsoDDE ofrece una base escalable para el diseño de fármacos con IA, proporcionando la fidelidad predictiva necesaria para navegar sistemas biológicos novedosos con una precisión sin precedentes.
Desde el lanzamiento de AlphaFold 3 en 2024 junto con Google DeepMind, el campo del descubrimiento de fármacos con IA ha avanzado a un ritmo extraordinario. Aunque AlphaFold 3 supuso un salto dramático en el rendimiento respecto a generaciones anteriores de modelos de predicción estructural, seguía siendo un desafío clave: comprender solo las estructuras biomoleculares no era suficiente para desbloquear programas reales de descubrimiento de fármacos in silico (en un ordenador).
El progreso en el diseño racional de fármacos —vital para resolver enfermedades humanas— requiere modelos predictivos altamente precisos, a lo largo de una amplia gama de propiedades e interacciones bioquímicas, capaces de trabajar en conjunto entre sí. Lo crucial es que, con tanto espacio biológico y químico aún sin explorar, estos modelos necesitan la capacidad de generalizar su poder predictivo más allá de sus conjuntos de entrenamiento hacia sistemas nuevos y invisibles.
Mientras seguimos abordando estos desafíos, nos complace presentar el Isomorphic Labs Drug Design Engine (IsoDDE) y presentar un pequeño conjunto de capacidades de IsoDDE a continuación y en nuestro informe técnico.
Predicción de la estructura de sistemas verdaderamente novedosos
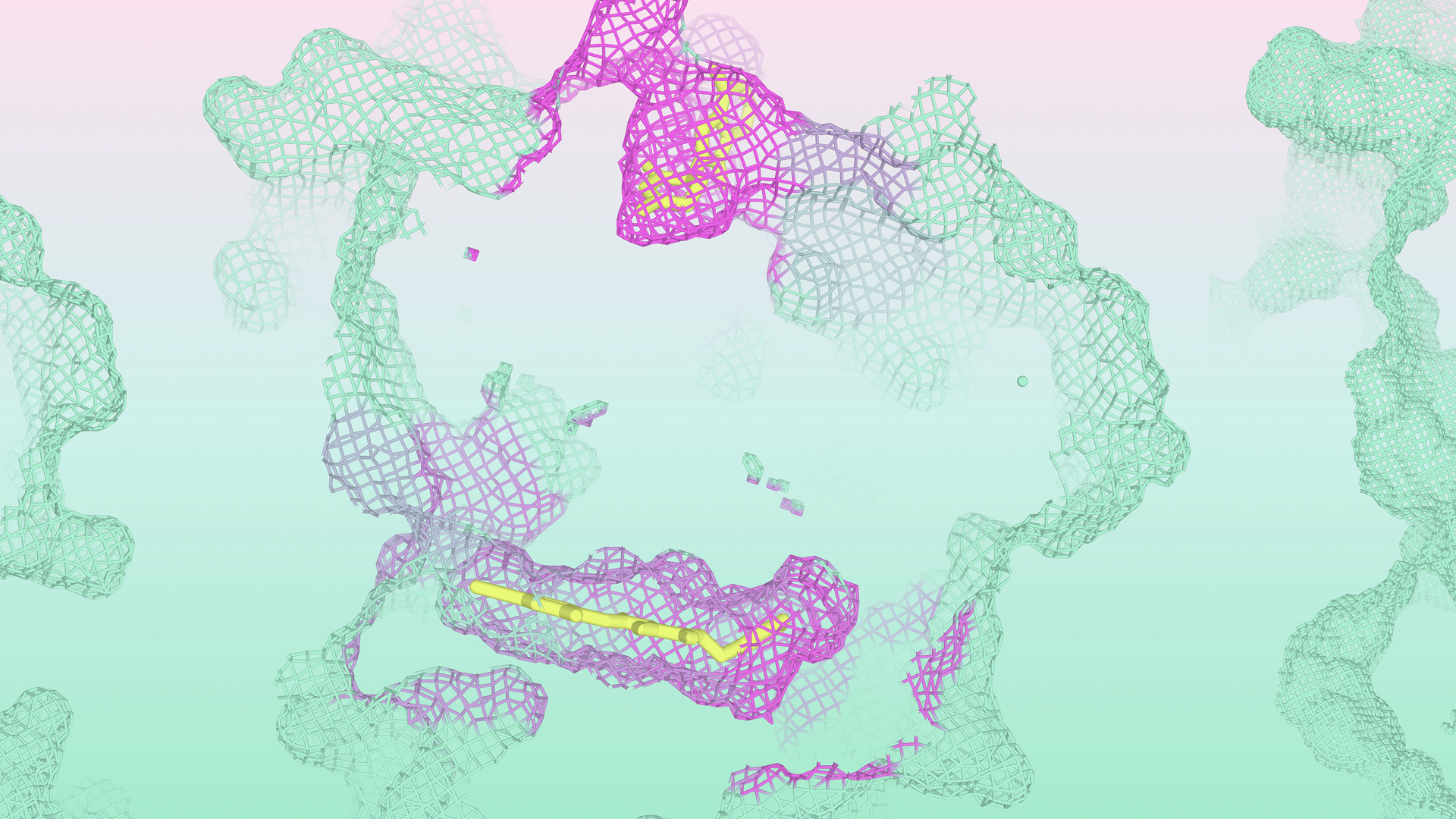
Predecir con precisión la estructura de las biomoléculas y cómo interactúan sigue siendo una capacidad crucial para el diseño racional de fármacos. Muchas tareas críticas posteriores se desbloquean al poder modelar con precisión los pequeños matices en la geometría de una proteína, ya sea entendiendo el impacto de mutaciones causantes de enfermedades o prediciendo qué moléculas se unirán a una proteína objetivo.
AlphaFold 3 transformó la predicción de la estructura proteína-ligando en el momento de su lanzamiento y la base de datos gratuita AlphaFold Protein Database aceleró la ciencia a una escala que hasta entonces era inimaginable. Hasta la fecha, ha sido utilizado por más de 3 millones de investigadores en más de 190 países.
Los benchmarks han revelado posteriormente que seguía existiendo una brecha en la precisión para estructuras diferentes a los ejemplos en los que se había entrenado AlphaFold 3. En otras palabras, que puede tener dificultades para generalizarse a regiones inexploradas del espacio biomolecular donde residen algunos de los mayores retos y oportunidades en el descubrimiento de fármacos.
IsoDDE demuestra un cambio progresivo en la capacidad de generalizarse a estructuras proteína-ligando que son muy diferentes a las de su conjunto de entrenamiento.
En el benchmark 'Runs N' Poses' (Škrinjar et al. 2025) —diseñado específicamente para probar la generalización a bolsillos y ligandos novedosos— IsoDDE más que duplica la precisión de AlphaFold 3 en los sistemas más difíciles.
Precisión de predicción de la estructura proteína-ligando en la categoría de generalización más difícil ((0-20]), la más diferente al conjunto de entrenamiento, según el benchmark 'Runs N' Poses'
En el informe, demostramos mediante varios ejemplos que podemos modelar con éxito eventos complejos y fuera de distribución, como los ajustes inducidos (donde una proteína adapta su forma para acomodar un ligando unido) y la apertura de bolsas crípticas (ocultas en ausencia de un ligando unido), mecanismos biológicos críticos, incluso cuando estos sistemas están alejados de los conjuntos de entrenamiento de tales modelos
Abriendo una nueva ventana para los biológicos complejos
Pero las moléculas pequeñas (como la aspirina) son solo una pieza del rompecabezas. A medida que las modalidades terapéuticas se expanden hacia biológicos complejos (como la insulina), la capacidad para modelar con precisión las interfaces anticuerpo-antígeno es fundamental.
IsoDDE proporciona un cambio progresivo en la precisión para este dominio. Supera a AlphaFold 3 en 2,3 veces y a Boltz-2 en 19,8 veces en el régimen de alta fidelidad (DockQ > 0,8) en un conjunto de pruebas de anticuerpos y antígenos novedoso y desafiante.
De forma crucial, IsoDDE muestra un rendimiento notable en el bucle CDR-H3 —la parte más variable y difícil de predecir de un anticuerpo—, desbloqueando efectivamente nuevas posibilidades para el diseño de anticuerpos de novo.
Precisión de la predicción de la estructura anticuerpo-antígeno para un conjunto de pruebas desafiante y de baja homología (n=334), mostrando la fracción de predicciones de alta calidad en la interfaz proteica (DockQ >0,8) a medida que aumenta el cálculo en tiempo de inferencia. IsoDDE supera a AlphaFold 3 en 2,3x y a Boltz-2 en 19,8x
Un nuevo estándar de oro para la predicción de afinidad de unión
Conocer la estructura 3D de un sistema bioquímico es solo el primer paso; La optimización eficaz de fármacos requiere saber cuán fuerte se unirá una molécula a su objetivo.
Los enfoques tradicionales están limitados al espacio químico similar a los datos de entrenamiento o por su alto coste computacional y dificultad de ejecución (por ejemplo, enfoques basados en física). Más recientemente han surgido métodos basados en aprendizaje profundo que aportan nueva velocidad a esta tarea, pero aún están por detrás de los enfoques basados en física en cuanto a precisión.
IsoDDE supera por un margen considerable a todos los métodos de aprendizaje profundo en tres benchmarks públicos: FEP+ 4, OpenFE y la reciente tarea de predicción de afinidad de enlace ciego CASP16.
De hecho, sorprendentemente, IsoDDE puede superar el rendimiento de métodos basados en física como el FEP, a pesar de que estos requieren fundamento en estructuras cristalinas experimentales y que IsoDDE no.
Al ofrecer predicciones de afinidad de unión altamente precisas a gran velocidad, IsoDDE permite a los investigadores clasificar y optimizar rápidamente moléculas potenciales a través de diversas series químicas durante los programas de diseño de fármacos.
Rendimiento de predicción de afinidad vinculante a través de una variedad de benchmarks públicos
Expansión del proteoma ligandable
La capacidad de identificar todos los bolsillos potenciales de una proteína, en ausencia de un ligando conocido, abre una serie de oportunidades únicas. Ya sea tratando con un objetivo de descubrimiento de fármacos de primer nivel sin anotación estructural o buscando una forma novedosa de modular una proteína bien estudiada, una capacidad general de identificación de bolsillos puede utilizarse para revelar el conjunto completo de posibles mecanismos de acción a seguir para el diseño molecular.
IsoDDE muestra la capacidad de identificar bolsillos nuevos y ligandoables incluso en ausencia de un ligando conocido y lejos del conjunto de entrenamiento del modelo. Esta capacidad para la identificación 'ciega' demuestra niveles de rendimiento que se acercan a técnicas experimentales como el remojo de fragmentos, que requieren grandes inversiones de tiempo, costes significativos y trabajo experimental en el mundo real. En comparación, IsoDDE se ejecuta en un ordenador en cuestión de segundos.
Podemos ver el poder de esta capacidad en el ejemplo del cereblon —un receptor de sustrato para el complejo ligasa CRL4 E3— que desempeña un papel clave en el marcado de proteínas dañadas o mal plegadas para la degradación proteasomal. Durante los últimos 15 años, se creía que existía una forma principal de drogar el cerebro: a través del clásico bolsillo de unión a la talidomida. Sin embargo, un estudio reciente (Dippon et al. 2026) descubrió experimentalmente un bolsillo de unión novedoso que era tanto alostérico (alejado del sitio tradicional de unión) como críptico (oculto en ausencia de un ligando de unión).
IsoDDE pudo recapitular el descubrimiento de este bolsillo, prediciendo la ubicación tanto de los sitios crípticos conocidos como de los novedosos usando solo la secuencia de cereblones como entrada, sin especificar la identidad de los ligandos. Además, una vez especificados los ligandos, IsoDDE pudo plegarlos correctamente en sus respectivos bolsillos en la orientación correcta.
IsoDDE pudo recapitular el reciente descubrimiento de un sitio críptico novedoso en cereblon (Dippon et al., 2026) que predecía la ubicación tanto de los sitios crípticos conocidos como de los novedosos usando solo la secuencia de cereblon como entrada, sin especificar la identidad de los ligandos.
Avanzando en el descubrimiento de fármacos
IsoDDE representa un salto adelante en precisión y capacidad, aportando una comprensión más profunda a las máquinas moleculares que componen el cuerpo humano y avanzando en el proceso de diseño de fármacos para modularlas.
Nuestros equipos dedicados al diseño de fármacos en Isomorphic Labs utilizan estas capacidades cada día en nuestros programas: para comprender estructuras invisibles, identificar bolsillos no caracterizados y crear materia química novedosa en la búsqueda de nuevos medicamentos para los pacientes.
Esperamos seguir explorando las fronteras del diseño de fármacos in silico y poner nuestras nuevas capacidades más potentes en objetivos de fármacos históricamente desafiantes.
Agradecemos a nuestros amigos de Google DeepMind por sus discusiones productivas y la colaboración.
Por favor, utiliza la siguiente cita si haces referencia a esta obra:
@manual{isodde2026,
title = {Accurate Predictions of Novel Biomolecular Interactions with IsoDDE},
author = {Isomorphic Labs Team},
month = feb,
year = 2026,
doi = {10.5281/zenodo.18606681},
url = {https://doi.org/10.5281/zenodo.18606681
Los pangolines son los mamíferos más traficados del mundo y, por ello, están expuestos a:
estrés extremo
contacto con múltiples especies
condiciones sanitarias deficientes
Todo esto los convierte en puntos calientes de intercambio viral.
Hasta ahora, la mayoría de estudios se habían centrado en pangolines enfermos, lo que distorsiona la imagen real de qué virus forman parte de su ecosistema natural. Este estudio corrige ese sesgo analizando pangolines sanos.
El viroma del pangolín saludable revela la diversidad viral en mamíferos y el riesgo zoonótico
El estudio analiza el viroma (todos los virus presentes) en pangolines sanos, para entender qué virus portan y qué riesgo tienen de saltar a humanos.
No trata sobre crear virus ni sobre ingeniería genética: es un estudio de ecología viral y riesgo zoonótico.
El objetivo es caracterizar todos los virus presentes en pangolines sanos, algo que casi no se había hecho. La mayoría de estudios previos se centraban en pangolines enfermos, lo que sesga la visión del “viroma” real de la especie.
Este tipo de investigación es ecología viral, no ingeniería genética.
Los autores realizaron:
Metatranscriptómica en 83 pangolines sanos
Comparación con datos previos de 52 pangolines enfermos
La metatranscriptómica permite detectar todos los virus activos en un organismo, sin necesidad de cultivarlos ni manipularlos.
👉 Es un método descriptivo, no experimental ni de ingeniería viral.
Objetivos del estudio
Identificar virus presentes en pangolines sanos.
Evaluar si esos virus están relacionados con virus que infectan humanos.
Estimar el riesgo de zoonosis (salto animal‑humano).
Metodología
Secuenciación metagenómica de muestras de pangolines.
Clasificación de virus encontrados (familias, géneros, similitud genética).
Comparación con virus conocidos en humanos y otros mamíferos
Gran diversidad viral
Encontraron múltiples familias de virus, incluyendo:
Coronaviridae
Paramyxoviridae
Picornaviridae
Herpesviridae
Otros virus típicos de mamíferos.
En 83 pangolines sanos identifican 51 vOTUs (unidades taxonómicas virales) pertenecientes a 6 familias de virus típicos de mamíferos:
Parvoviridae
Picornaviridae
Papillomaviridae
Circoviridae
Flaviviridae
Paramyxoviridae
👉 Nada de coronavirus tipo SARS‑CoV‑2.
Coronavirus relacionados con otros virus animales
Detectaron coronavirus relacionados con virus de murciélagos y otros mamíferos, pero no SARS‑CoV‑2 ni un precursor directo.
Evidencia de que los pangolines pueden actuar como hospedadores intermedios
El estudio sugiere que los pangolines pueden portar virus con potencial zoonótico, pero no afirma que sean el origen del SARS‑CoV‑2.
Comparación con pangolines enfermos
Usan datos previos de 52 pangolines enfermos y encuentran:
Algunos virus solo aparecen en los enfermos, como Orthopneumovirus hominis y Orthorubulavirus mammalis.
Esto sugiere que podrían estar implicados en la enfermedad, pero no es una prueba definitiva
Esto los convierte en candidatos a virus patógenos en pangolines.
Evidencia de recombinación y transmisión entre especies
Detectaron recombinación en virus de la familia Paramyxoviridae entre:
pangolines
perros domésticos
Esto indica flujo viral entre especies, algo típico en animales traficados o en contacto con humanos.
Co‑infecciones relevantes
Encuentran correlación fuerte entre dos parvovirus:
Copiparvovirus P171T/pangolin/2018
Pangolin protoparvovirus
Esto sugiere que podrían transmitirse juntos o compartir rutas de contagio.
Esto sugiere rutas de transmisión compartidas o sinergias entre virus.
Evaluación del riesgo zoonótico
Identifican 16 virus con potencial de infectar humanos, entre ellos:
Pangolin pestivirus
Manis javanica papillomavirus 1
👉 Esto NO significa que vayan a causar una pandemia.
Significa que tienen características que merecen vigilancia.
Lo que sí aporta el estudio
Un mapa del viroma natural del pangolín.
Identificación de virus que podrían saltar a humanos.
Evidencia de transmisión entre especies en contextos de tráfico ilegal.
Bases para programas de vigilancia epidemiológica
Lo que NO implica
No estudia coronavirus tipo SARS‑CoV‑2.
No describe manipulación genética.
No realiza experimentos de ganancia de función.
No sugiere creación de virus en laboratorio.
No aporta evidencia sobre el origen del COVID‑19.
Es un estudio de ecología viral, no de ingeniería virológica.
No hay manipulación genética
Es un estudio puramente descriptivo
El estudio encaja en una línea de investigación común:mapear virus en animales salvajes para anticipar riesgos de zoonosis.
Este tipo de trabajos:
ayudan a identificar virus peligrosos antes de que salten a humanos
no implican creación ni modificación de virus
no aportan evidencia sobre el origen del SARS‑CoV‑2
Este artículo no contiene nada que sugiera creación o modificación de SARS‑CoV‑2.
Background Pangolins, the world’s most trafficked mammals, have emerged as critical subjects of study due to their potential role as intermediate hosts for zoonotic viruses. While previous studies have primarily focused on diseased pangolins, the virome composition of healthy individuals remains largely unexplored. Results To address this knowledge gap, we performed comprehensive metatranscriptomic analysis of 83 healthy pangolins, in comparison with virome data of 52 diseased individuals derived from previously published datasets. We identified 51 viral operational taxonomic units (vOTUs) across six mammalian-associated viral families: Parvoviridae, Picornaviridae, Papillomaviridae, Circoviridae, Flaviviridae, and Paramyxoviridae. Notably, we observed recombination in Morbillivirus canis isolate BJ16B35, Canine distemper virus strain PS, and UN_MBA191024-Paramyxoviridae-1 from pangolins and domestic dogs, suggesting cross-species transmission dynamics. Co-infection analysis revealed a strong positive correlation between Copiparvovirus P171T/pangolin/2018 and Pangolin protoparvovirus, suggesting possible shared transmission pathways. Several viruses, including Orthopneumovirus hominis and Orthorubulavirus mammalis, were exclusively detected in diseased pangolins, implicating their potential role in pathogenesis. Zoonotic risk assessment identified 16 vOTUs with high predicted potential for human infection, including Pangolin pestivirus and Manis javanica papillomavirus 1. Conclusions Our findings significantly expand our understanding of viral diversity in healthy pangolins and help distinguish commensal viral communities from potentially pathogenic ones. This research underscores the importance of continued wildlife viral surveillance for both conservation and public health preparedness
En conclusión, este estudio avanza significativamente en nuestra comprensión del viroma del pangolín y su relevancia para la vigilancia zoonótica. Al establecer perfiles virales de referencia en individuos saludables e identificar candidatos de alto riesgo, proporcionamos una base para un monitoreo y evaluación de riesgos más específicos. A medida que la vida silvestre continúa enfrentando presiones ambientales sin precedentes, la caracterización proactiva del viroma puede emplearse como una estrategia crítica en la preparación para la salud global, ofreciendo información importante sobre el panorama viral antes de que ocurran eventos de transmisión entre especies.
Documentos obtenidos por FOIA muestran que participó como investigador en proyectos financiados por agencias de EE. UU. (NIAID, USAID) sobre coronavirus, algunos catalogados como de “gain of function” por críticos
En algunos casos, modificar virus para estudiar qué cambios aumentan infectividad o tropismo - “gain of function”-
Es cierto que el WIV trabajaba con coronavirus de murciélagos cercanos a SARS‑CoV‑2 y que estudiaba qué mutaciones podían aumentar su capacidad de infectar humanos.
También es cierto que muchos virólogos consideran plausible que un virus de murciélago haya pasado a humanos mediante la cadena de comercio de fauna salvaje, sin necesidad de manipulación de laboratorio
Hasta ahora, no se ha publicado ningún constructo de laboratorio que sea el “padre” directo de SARS‑CoV‑2, ni una secuencia previa que encaje como “prototipo” diseñado. La comunidad científica, incluso quienes ven posible una fuga de laboratorio, reconoce que falta esa pieza clave
Posibilidades
Origen natural (zoonosis): Muchos estudios apuntan al mercado de Huanan y a la cadena de fauna salvaje como foco temprano, con varios linajes del virus circulando ya en diciembre de 2019. Esto encaja bien con un salto animal‑humano, aunque aún no se ha identificado el animal intermedio
Fuga de laboratorio: Algunos organismos de inteligencia de EE. UU. consideran “más probable” una fuga de laboratorio, pero con baja confianza y sin pruebas directas.
Otros organismos se inclinan por origen natural.
En conjunto, los propios informes oficiales reconocen que no hay prueba definitiva para ninguna de las dos hipótesis
Tipo de trabajos científicos sí son relevantes para el debate sobre el origen del SARS‑CoV‑2
Estudios sobre coronavirus de murciélagos cercanos a SARS‑CoV‑2
Estos son los trabajos más relevantes porque:
describen virus similares al SARS‑CoV‑2
analizan su capacidad de infectar células humanas
estudian su evolución, recombinación y tropismo
Ejemplos de estudios relevantes (en general, no solo de Hu):
Secuenciación de coronavirus tipo SARS‑like
Trabajos que describen virus como:
RaTG13
BANAL‑52
RmYN02
SHC014
WIV1
Estos virus comparten entre 90–96% de identidad con SARS‑CoV‑2 y son claves para entender su origen.
✔️ Estudios de recombinación en coronavirus
Porque SARS‑CoV‑2 tiene señales de recombinación natural en su historia evolutiva.
Estudios de unión al receptor ACE2
Porque la afinidad del virus por ACE2 humano es una de sus características más llamativas.
Trabajos que implican cultivos virales, quimeras o experimentos de tropismo
Aquí es donde entra la parte más delicada del debate.
Estos estudios son relevantes porque:
manipulan coronavirus para estudiar su infectividad
crean virus quiméricos (combinando partes de distintos virus
prueban qué mutaciones aumentan la capacidad de infectar humanos
Este tipo de investigación no es evidencia de creación de SARS‑CoV‑2, pero sí es relevante porque:
muestra qué técnicas se usaban en ciertos laboratorios
indica qué virus estaban siendo manipulados
ayuda a evaluar si una fuga accidental es plausible
Ejemplos típicos (de distintos grupos de investigación):
✔️ Experimentos con SHC014 y WIV1
Estos son los famosos estudios que generaron virus quiméricos para probar infectividad en células humanas.
Propuestas de investigación como DEFUSE (no ejecutadas)
La propuesta de EcoHealth a DARPA incluía insertar sitios de corte tipo furina en coronavirus.
DARPA no la financió, pero se cita mucho en el debate.
Estudios epidemiológicos y genómicos sobre los primeros casos humanos
Estos trabajos no tienen que ver con laboratorios, pero sí con el origen del virus.
Incluyen:
análisis de los primeros linajes (A y B)
estudios del mercado de Huanan
reconstrucciones filogenéticas
análisis de mutaciones tempranas
estudios de serología previa a diciembre de 2019
Estos trabajos ayudan a responder:
¿circulaba el virus antes del brote oficial?
¿hubo múltiples introducciones?
¿hay evidencia de un animal intermedio?
🧩 ¿Qué trabajos de Ben Hu entran en estas categorías?
Aquí es donde se aclara mucho el panorama:
✔️ Relevantes para el debate
Los trabajos de Hu que estudian:
coronavirus de murciélagos tipo SARS‑like
infectividad en células humanas
tropismo viral
caracterización de proteínas spike
estudios de recombinación en coronavirus
Estos sí son relevantes porque se alinean con las líneas de investigación que podrían, en teoría, generar virus con propiedades parecidas a SARS‑CoV‑2.
No relevantes para el debate
Trabajos como:
estudios de viroma de pangolines
estudios de virus no relacionados con coronavirus
análisis ecológicos de fauna salvaje
papers sobre virus que no usan ACE2
Estos no aportan nada al debate sobre el origen del COVID, aunque a veces se citan erróneamente solo porque Hu aparece como coautor.
En resumen
Los trabajos relevantes para el debate sobre el origen del SARS‑CoV‑2 son aquellos que:
estudian coronavirus muy similares al SARS‑CoV‑2
analizan su capacidad de infectar humanos
implican manipulación experimental de coronavirus
reconstruyen la evolución temprana del virus en humanos
Los estudios de viroma general (como el de pangolines que analizamos antes) no tienen relación directa con este debate
El hígado es un órgano multifuncional. Elimina toxinas de la sangre, produce bilis para la digestión y combate infecciones, entre cientos de funciones vitales. Sin embargo, exponerse a estos factores tiene un precio: la exposición prolongada a toxinas y al alcohol, pueden provocar daños y la formación de tejido cicatricial, conocido como fibrosis.
En un nuevo estudio, investigadores examinaron el hígado de un ratón, primero haciendo que el tejido sea semitransparente. Después, con partículas fluorescentes que se depositan en diferentes estructuras, como el conducto biliar (verde) y la vena hepática (amarilla), se identificaron lugares donde las partículas verdes se filtran y se acumulan en el tejido circundante (círculo en rojo). Estas estructuras recién descubiertas, a las que denominan complejo lamelar periportal, podrían estar implicadas en los cambios que sufre el tejido hepático durante la fibrosis y ser un nuevo objetivo para fármacos
The liver is a multipurpose organ. It cleans toxins out of the blood, produces bile that helps with digestion, and helps to fight against infections, among hundreds of vital jobs. But putting itself in harm’s way comes at a price – prolonged exposure to toxins like alcohol can lead to damage and scar tissue called fibrosis. Here researchers zoom in on a mouse’s liver with an advanced microscope, first rendering the tissue semi-transparent by clearing away chemicals that block their view. Using fluorescent particles that settle in different structures like the bile duct (green) and hepatic vein (yellow), the team spot places where the green particles 'leak' and pool in the surrounding tissue. These newly discovered structures, which they call the periportal lamellar complex, may be involved in changes to the liver tissue during fibrosis and a future target for medical drugs.
Four-way collaboration brings together world-leading AI and biological expertise to make AI-predicted protein complex structures openly available to the global scientific community
Slime mold Dictyostelium discoideum protein complex Q55DI5 (AF-0000000066503175), annotated as a transcription elongation factor. The single chain looks disordered, but the homodimer reveals that two chains intertwine, each contributing half a domain to form a stable fold. An illustration of how predicting protein complexes reveals biology that single-protein models miss. Credit: AlphaFold Database, background by Karen Arnott/EMBL-EBI
A new collaboration between EMBL’s European Bioinformatics Institute (EMBL-EBI), Google DeepMind, NVIDIA, and Seoul National University has made millions of AI-predicted protein complex structures openly available through the AlphaFold Database. To maximise global health impact, the dataset prioritises proteins important for understanding human health and disease. This is the largest dataset of protein complex predictions currently available.
Proteins are the building blocks of life. They interact to create protein complexes which fulfil biological functions. By visualising protein interactions, scientists can uncover the molecular mechanisms that drive cell behaviour, identify what goes wrong when someone gets sick, and develop new drugs and therapies. Predicting the structure of protein complexes is extremely challenging because, in nature, proteins change shape and interact in many different ways.
“Science thrives on collaboration,” said Jo McEntyre, Interim Director of EMBL-EBI. “By making this foundational protein complex dataset openly available to the world, we’re inviting researchers to test, refine, and build on it to drive the next wave of biological discoveries.”
Protein complexes for global health impact
The latest AlphaFold Database update spans millions of homodimers – protein complexes formed of two identical proteins. It focuses on 20 of the most studied species, including humans, as well as the World Health Organization’s priority pathogens list. This approach aims to bring significant and immediate value for global health challenges.
“By expanding the AlphaFold Database to include protein complexes, we are addressing a critical need expressed by the scientific community,” said Anna Koivuniemi, Head of the Google DeepMind Impact Accelerator. “We hope that by lowering the barrier to these complex predictions, we can empower researchers everywhere to pursue the next wave of discoveries that could ultimately improve human health on a global scale.”
Scientific expertise meets technical innovation
The collaboration builds on Google DeepMind’s AI system AlphaFold, which, since 2021, accurately predicted the structure of millions of proteins. To democratise access to AlphaFold predictions, Google DeepMind and EMBL-EBI developed the AlphaFold Database, an open resource that anyone can access. The database has over 3.4 million users from 190 countries.
Through ongoing dialogue with the scientific community, a clear need emerged to expand the AlphaFold database to include protein complexes. In response to this need, EMBL-EBI, Google DeepMind, NVIDIA, and Seoul National University teamed up, contributing specialist expertise and resources, to calculate and integrate millions of protein complexes into the AlphaFold Database.
“By making this foundational dataset openly available to the world, we’re inviting researchers to test, refine, and build on it to drive the next wave of biological discoveries.”
The collaboration brought together deep biological expertise and technical innovations. NVIDIA and the Steinegger Lab at the Seoul National University developed the methodology, based on Google DeepMind’s AI system AlphaFold, including accelerations to multiple sequence alignment calculations and deep learning inference. NVIDIA provided cutting-edge AI infrastructure and scaled out inference pipelines to overcome limitations that historically made this scale of calculations challenging. EMBL-EBI enabled the collaboration by bringing the other parties together and contributing expertise in scientific and biodata management, as well as analysis. As a champion of open science, EMBL-EBI, together with Google DeepMind, integrated the new dataset into the AlphaFold Database.
“NVIDIA’s ambition is to consistently contribute orders-of-magnitude accelerations for fundamental digital biology workloads, enabling what was not possible before,” said Anthony Costa, NVIDIA Director of Digital Biology. “This release is a great example of how AI infrastructure and software can uniquely enable new scales of biological understanding.”
“By making predicted protein complexes accessible at an unprecedented scale, we are illuminating an unseen landscape of molecular interactions across the tree of life,” explained Martin Steinegger, Associate Professor at Seoul National University.
Open science at scale
It takes a blend of AI-scale infrastructure and deep technical knowledge in accelerating complex workflows to generate AI predictions for protein complexes at this scale. The collaboration is centrally hosting data that would otherwise require around 17 million hours of GPU (graphics processing unit) computing to recreate.
By making these calculations once and adding the information into the AlphaFold Database, this collaboration aims to help democratise access to protein complex predictions. It enables scientists everywhere to investigate how proteins interact in the vast protein universe, and accelerate discoveries that could lead to new medicines, new products, and a deeper understanding of life itself.
“This release is a great example of how AI infrastructure and software can uniquely enable new scales of biological understanding.”
This is the first step in an ambition to add a wide range of protein complex structure predictions to the AlphaFold Database. The partnership has already calculated predictions for 30 million complexes. Of these, 1.7 million high-confidence homodimer predictions have been added to the AlphaFold Database. Another 18 million are lower-confidence homodimers, which will be made available as a list and for bulk download from the EMBL-EBI FTP server in the coming days. The rest are heterodimers, currently being analysed and assessed. More protein complex predictions will be calculated and high-confidence predictions will be added to the AlphaFold Database in the coming months. The work is described in more detail in this preprint.
“The human genome has just over 20,000 different proteins. Despite this relatively small genome, human beings display incredibly complex pathways, processes and regulation. Much of this complexity arises from the intermolecular interactions between proteins, and with small molecule ligands and DNA. Adding predicted protein-protein homodimeric interactions to the AlphaFold Database is a first step towards a comprehensive description of the human interactome, the basis by which human biology will be described and understood. This has relevance for the design of new therapeutics, understanding host-pathogen interactions, and more. Making these structures accessible to all, allows every researcher around the world to build on these data, moving one step closer to predicting the biology of life,” said Dame Janet Thornton, Director Emeritus of EMBL-EBI.
Milioni di complessi proteici aggiunti al Database Alphafold fanno luce su come le proteine interagiscono
Una collaborazione internazionale mette insieme le principali competenze mondiali nell’AI e nella biologia per rendere accessibili alla comunità scientifica globale le predizioni delle strutture dei complessi proteici.
Una nuova collaborazione tra l’Istituto di Bioinformatica dell’EMBL (EMBL-EBI), Google DeepMind, NVIDIA, e la Seoul National University, ha reso accessibili le predizioni basate sull’AI di milioni si strutture di complessi proteici attraverso il Database Alphafold. Per massimizzare l’impatto sulla salute globale, il dataset ha dato priorità alle proteine importanti per la comprensione della salute umana e delle malattie. Questo è il più grande dataset attualmente disponibile di previsioni sui complessi proteici.
Le proteine sono i mattoni della vita. Interagiscono tra loro per formare complessi proteici che svolgono funzioni biologiche. Visualizzando le interazioni tra proteine, gli scienziati possono scoprire i meccanismi molecolari che guidano il comportamento delle cellule, identificare cosa non funziona quando una persona si ammala e sviluppare nuovi farmaci e terapie. Prevedere la struttura dei complessi proteici è estremamente difficile perché, in natura, le proteine cambiano forma e interagiscono in molti modi diversi.
“La scienza progredisce attraverso la collaborazione”, ha detto Jo McEntyre, Direttrice ad Interim dell’EMBL-EBI. “Rendendo questo dataset di complessi proteici disponibile alla comunità scientifica, invitiamo i ricercatori a testarlo, perfezionarlo e svilupparlo ulteriormente, per guidare la prossima ondata di scoperte biologiche”.
Complessi proteici per un impatto sulla salute globale
L’ultimo aggiornamento del Database Alphafold comprende milioni di omodimeri – complessi proteici formati da due proteine identiche. Si concentra su 20 delle specie più studiate, tra cui l’uomo, oltre che sulla lista dei batteri patogeni considerati prioritari dall’Organizzazione Mondiale della Sanità. Questo approccio mira ad avere un impatto significativo e immediato per affrontare le sfide della salute globale.
“Espandere il Database Alphafold con l’aggiunta dei complessi proteici risponde ad un’esigenza critica espressa dalla comunità scientifica”, ha detto Anna Koivuniemi, a capo dell’Impact Accelerator di Google DeepMind. “Ci auguriamo che, rendendo accessibili queste informazioni, consentiremo ai ricercatori di tutto il mondo di guidare la prossima ondata di scoperte, che potrebbero migliorare la salute umana su scala globale”.
La collaborazione si basa sul sistema di AI sviluppato da Google DeepMind – Alphafold – che dal 2021 ha previsto con elevata precisione la struttura di milioni di proteine. Per democratizzare l’accesso alle previsioni di Alphafold, Google DeepMind e EMBL-EBI hanno sviluppato il Database Alphafold, una risorsa aperta accessibile a chiunque. Il database conta oltre 3,4 milioni di utenti provenienti da 190 paesi.
Attraverso un dialogo continuo con la comunità scientifica, è emersa la necessità di espandere il Database Alphafold per includere anche i complessi proteici. In risposta a questa esigenza, EMBL-EBI, Google DeepMind, NVIDIA e la Seoul National University hanno unito le forze, contribuendo con competenze specifiche e risorse per calcolare e integrare milioni di complessi proteici nel Database Alphafold.
“Rendendo questo dataset di complessi proteici disponibile alla comunità scientifica, invitiamo i ricercatori a testarlo, perfezionarlo e svilupparlo ulteriormente, per guidare la prossima ondata di scoperte biologiche”
NVIDIA e il gruppo di Steinegger alla Seoul National University hanno sviluppato la metodologia, basata sul sistema di AI di Google DeepMind Alphafold, includendo accelerazioni nei calcoli di allineamento di sequenze multiple e nelle previsioni di deep learning. NDIVIA ha fornito infrastrutture di AI all’avanguardia e ha ottimizzato le pipeline di calcolo per superare i limiti che storicamente rendevano difficile eseguire calcoli su larga scala. EMBL-EBI ha reso possibile la collaborazione riunendo le diverse parti e contribuendo con competenze nella gestione e nell’analisi dei dati scientifici e biologici. Come sostenitore della open science, EMBL-EBI insieme a Google DeepMind ha integrato il nuovo dataset nel Database Alphafold.
“L’ambizione di NVIDIA è di fornire costantemente accelerazioni di ordini di grandezza per i processi fondamentali della biologia digitale, permettendo ciò che prima non era possibile”, ha dichiarato Anthony Costa, Direttore di Digital Biology di NVIDIA. “Questa espansione del Database è un ottimo esempio di come infrastruttura e software basati su AI possano raggiungere nuovi livelli di comprensione scientifica”. “Rendendo le previsioni sui complessi proteici accessibili su una scala senza precedenti, stiamo illuminando un panorama fino ad ora invisibile di interazioni molecolari lungo l’albero della vita”, ha spiegato Martin Steinegger, Professore Associato presso la Seoul National University.
Open science su larga scala
Per accelerare processi complessi e generare previsioni AI sui complessi proteici, è necessario un mix di infrastrutture AI su larga scala e accurata conoscenza tecnica. La collaborazione ospita centralmente dati che, altrimenti, richiederebbero circa 17 milioni di ore di calcolo GPU (graphics processing unit) per essere ricreati.
Rendendo questi calcoli disponibili una sola volta e integrando le informazioni nel Database Alphafold, la collaborazione mira a democratizzare l’accesso alle previsioni sui complessi proteici. Questo permette agli scienziati di tutto il mondo di studiare come le proteine interagiscono nell’immenso universo biologico e di accelerare scoperte che potrebbero portare a nuovi farmaci, nuovi prodotti e a una comprensione più profonda della vita stessa.
“Questo risultato è un ottimo esempio di come infrastruttura e software basati su AI possano raggiungere nuovi livelli di comprensione scientifica”
Questo è il primo passo verso l’ambizione più ampia di includere una vasta gamma di previsioni di strutture di complessi proteici al Database Alphafold. La partnership ha già calcolato previsioni per 30 milioni di complessi. Di questi, 1,7 milioni di previsioni di omodimeri ad alta affidabilità sono state aggiunte al Database Alphafold. Altre 18 milioni di strutture di omodimeri, con affidabilità più bassa, sono disponibili come liste per il download in blocco. Il resto delle strutture sono eterodimeri, attualmente in fase di analisi e valutazione. Ulteriori previsioni di complessi proteici saranno calcolate e quelle ad alta affidabilità saranno integrate nel Database Alphafold nei prossimi mesi. Il lavoro è descritto più in dettaglio in un preprint.
“Il genoma umano contiene poco più di 20.000 proteine diverse. Nonostante questo genoma relativamente piccolo, gli esseri umani mostrano percorsi, processi e regolazioni incredibilmente complessi. Gran parte di questa complessità deriva dalle interazioni intermolecolari tra proteine, ligandi di piccole molecole e DNA. Aggiungere le previsioni di interazioni proteina-proteina omodimeriche al Database Alphafold è un primo passo verso una descrizione completa dell’interattoma umano, la base su cui sarà descritta e compresa la biologia umana. Questo ha rilevanza per progettare nuovi farmaci, comprendere le interazioni ospite-patogeno e molto altro. Rendere queste strutture accessibili a tutti permette a ogni ricercatore nel mondo di basarsi su questi dati, e di avvicinarsi sempre di più alla predizione della biologia della vita”, ha dichiarato Dame Janet Thornton, Direttrice Emerita di EMBL-EBI