A Python library for probabilistic analysis of single-cell omics data
Nature Biotechnology (2022)
A pesar del atractivo de los modelos probabilísticos, varios obstáculos impiden su adopción por parte de la comunidad. El primer obstáculo, que proviene de la perspectiva del usuario final, está relacionado con la dificultad de implementar y ejecutar dichos modelos. Dado que los modelos probabilísticos suelen implementarse utilizando bibliotecas de aprendizaje automático de Python, los usuarios suelen tener que interactuar con interfaces y objetos de menor nivel que los utilizados en entornos populares para el análisis de datos unicelulares como Bioconductor6, Seurat7 o Scanpy8.
Un segundo obstáculo está relacionado con el desarrollo de nuevos modelos probabilísticos. Desde el punto de vista de los desarrolladores, son muchas las rutinas necesarias que hay que implementar en apoyo de un modelo probabilístico, incluyendo el manejo de datos, los cálculos de tensor, las rutinas de entrenamiento que manejan la gestión de dispositivos (por ejemplo, la computación de la GPU (unidad de procesamiento gráfico)), y la optimización subyacente, el muestreo y los procedimientos numéricos. Aunque los paquetes de aprendizaje automático de alto nivel que automatizan algunas de estas rutinas (por ejemplo, PyTorch Lightning9 o Keras10) son cada vez más populares, no funcionan sin problemas con los datos ómicos unicelulares.
Para abordar estas limitaciones, presentamos scvi-tools (https://scvi-tools.org/), una biblioteca de Python para el análisis probabilístico profundo de datos ómicos unicelulares. Desde la perspectiva del usuario final (Nota Suplementaria 1), scvi-tools ofrece acceso estandarizado a métodos para muchas tareas de análisis de datos unicelulares, como la integración de datos de secuenciación de ARN unicelular (scRNA-seq) (scVI11 o scArches12), la anotación de perfiles unicelulares (CellAssign13 o scANVI14), la deconvolución de perfiles transcriptómicos espaciales masivos (Stereoscope15 o DestVI16), la detección de dobletes (Solo17) y el análisis multimodal de datos CITE-seq (indexación celular de transcriptomas y epítopos por secuenciación) (totalVI18)
En el proceso de análisis más amplio, scvi-tools se encuentra después del preprocesamiento inicial basado en el control de calidad (QC) y genera resultados que pueden ser interpretados a través de herramientas generales de análisis de células individuales (Fig. 1a). En su núcleo, scvi-tools implementa varias funcionalidades clave que son accesibles a través de las modalidades de datos, como el análisis diferencial y la integración de conjuntos de datos. Los 14 modelos (Tabla Suplementaria 1) actualmente implementados en scvi-tools interactúan con Scanpy a través del formato de conjunto de datos anotados (AnnData19), y los modelos comparten una interfaz de usuario consistente (Fig. 1b). La biblioteca de scvi-tools también tiene una interfaz con R, de manera que cada modelo puede ser utilizado en las tuberías de Seurat o Bioconductor.

a, Overview of single-cell omics analysis pipeline with scvi-tools. Datasets may contain multiple layers of omic information, along with metadata at the cell and feature levels. QC and preprocessing are done with popular packages like Scanpy, Seurat and Scater. Subsequently, datasets can be analyzed with scvi-tools, which contains implementations of probabilistic models that offer a range of capabilities for various omics. Finally, results are further investigated or visualized, typically through a nearest neighbors graph, and through environments like VISION or cellxgene or by directing back to Scanpy or Seurat. b, Left, overview of the functionality of models implemented in scvi-tools covers core single-cell analysis tasks. Right, each model has a simple and consistent user interface; the code snippet shown applies scVI to a dataset read from a h5ad file and then performs dimensionality reduction and differential expression.
Desde la perspectiva de un desarrollador de métodos, scvi-tools ofrece un conjunto de bloques de construcción que facilitan la implementación de nuevos modelos y la modificación de modelos existentes con una mínima sobrecarga de código (Fig. 2a,b y Nota Suplementaria 4). Estos bloques de construcción utilizan librerías populares, como AnnData12, PyTorch20, PyTorch Lightning9 y Pyro21, y facilitan el diseño de modelos probabilísticos con componentes de redes neuronales y aceleración en la GPU. Esto permite a los desarrolladores de métodos centrarse principalmente en el desarrollo de modelos probabilísticos en lugar de en la gestión de datos, el entrenamiento del modelo y el código de la interfaz de usuario. Demostramos cómo estos bloques de construcción pueden utilizarse para el desarrollo eficiente de modelos a través de una reimplementación de Stereoscope, en la que demostramos una reducción sustancial de la complejidad del código (Fig. 2c-e y Nota complementaria 5). Este ejemplo demuestra el amplio alcance de los análisis que pueden ser impulsados por scvi-tools.
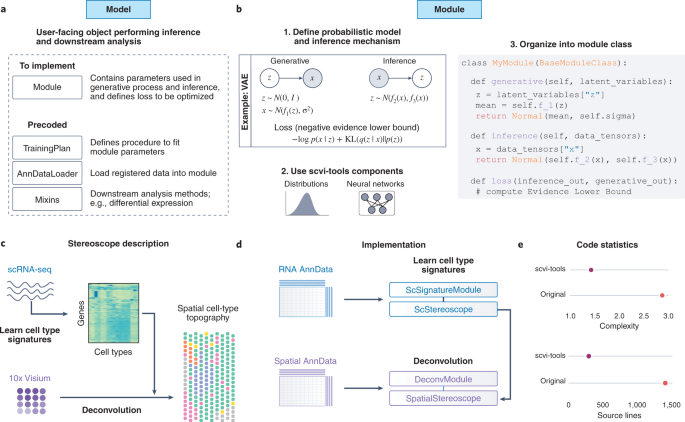
a, For every probabilistic method implemented in scvi-tools, users interact with a high-level ‘model’ object. The model relies on several lower level components for training a model and analyzing data. The ‘module’, which must be implemented, systematically encapsulates the probabilistic specification of the method. The rest of the lower level components rely on precoded objects in scvi-tools, such as AnnDataLoader for loading data from AnnData objects, TrainingPlan for updating the parameters of the module, and Mixin classes for downstream analyses. b, The creation of a new module in scvi-tools involves three key steps. First, the generative model and inference procedure are mathematically specified. Second, users may either choose from our wide range of precoded neural network architectures and distributions or implement their own with PyTorch. Finally, those elements are combined together and organized into a class that inherits from the abstract class BaseModuleClass (note: presentation is pseudocode). The generative method maps latent variables to the data-generating distribution. The inference method maps input data to the variational distribution (specific to variational inference). The loss method specifies the objective function for the training procedure, here the evidence lower bound (and specifically depicted for a variational autoencoder (VAE)). c, Overview of the Stereoscope method. Stereoscope takes as input a spatial transcriptomics dataset, as well as a single-cell RNA sequencing dataset, and outputs the proportion of cell types in every spot. d, Short description of the steps required to reimplement Stereoscope into the codebase. For each of the two models of Stereoscope, we created a module class as well as a model class. e, Average cyclomatic code complexity and total number of source code lines for each of scvi-tools implementation and the original implementation.
En el desarrollo de scvi-tools, nuestro objetivo es cerrar la brecha que existe entre el ecosistema de software unicelular y los marcos contemporáneos de aprendizaje automático para construir y desplegar esta clase de modelos. Así, los desarrolladores pueden ahora esperar construir modelos que son inmediatamente accesibles a los usuarios finales en la comunidad de células individuales, mientras continúan confiando en las bibliotecas populares de aprendizaje de máquinas. En nuestro sitio web de documentación, ofrecemos una serie de tutoriales sobre la construcción de un modelo con scvi-tools, recorriendo los pasos de la gestión de datos, la construcción de módulos y el desarrollo del modelo. También hemos creado un repositorio de plantillas en GitHub que permite a los desarrolladores crear rápidamente un paquete de Python que utiliza pruebas unitarias, documentación automatizada y bibliotecas populares de estilo de código. Este repositorio demuestra cómo se pueden utilizar los bloques de construcción de scvi-tools para el despliegue de modelos externos. Anticipamos que la mayoría de los modelos construidos con scvi-tools se desplegarán de esta manera como paquetes independientes mientras se adhieren a la API estándar y a las convenciones de codificación, lo que los hará más fácilmente accesibles para los nuevos usuarios.
Como scvi-tools sigue en desarrollo activo, los usuarios finales pueden esperar que scvi-tools evolucione continuamente, añadiendo soporte para nuevos modelos, nuevos flujos de trabajo y nuevas características. Anticipamos que estos recursos servirán a la comunidad unicelular facilitando la creación de prototipos de nuevos modelos, creando un estándar para el despliegue de software de análisis probabilístico y mejorando el proceso de descubrimiento científico.
References
Svensson, V., da Veiga Beltrame, E. & Pachter, L. Database 2020, baaa073 (2020).
Lee, J., Hyeon, D. Y. & Hwang, D. Exp. Mol. Med. 52, 1428–1442 (2020).
Wagner, A., Regev, A. & Yosef, N. Nat. Biotechnol. 34, 1145–1160 (2016).
Zappia, L., Phipson, B. & Oshlack, A. PLOS Comput. Biol. 14 (2018).
Lopez, R., Gayoso, A. & Yosef, N. Mol. Syst. Biol. 16, e9198 (2020).
Gentleman, R. C. et al. Genome Biol. 5, R80 (2004).
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Nat. Biotechnol. 33, 495–502 (2015).
Wolf, F. A., Angerer, P. & Theis, F. J. Genome Biol. 19, 15 (2018).
Falcon, W. & The PyTorch Lightning team. PyTorch Lightning (Version 1.4). (2019); https://doi.org/10.5281/zenodo.3828935
Chollet, F. et al. Keras. https://keras.io (2015).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Nat. Methods 15, 1053–1058 (2018).
Lotfollahi, M. et al. Nat. Biotechnol. 40, 121–130 (2022).
Zhang, A. W. et al. Nat. Methods 16, 1007–1015 (2019).
Xu, C. et al. Mol. Syst. Biol. 17, e9620 (2021).
Andersson, A. et al. Commun. Biol. 3, 565 (2020).
Lopez, R. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.05.10.443517 (2021).
Bernstein, N. J. et al. Cell Syst. 11, 95–101.e5 (2020).
Gayoso, A. et al. Nat. Methods 18, 272–282 (2021).
Angerer, P., Wolf, A., Virshup, I. & Rybakov, S. AnnData. GitHub https://github.com/theislab/anndata (2019).
Paszke, A. et al. Adv. Neural Inf. Process. Syst. 32, 8026–8037 (2019).
Bingham, E. et al. J. Mach. Learn. Res. 20, 1–6 (2019).
Acknowledgements
We acknowledge members of the Streets and Yosef laboratories for general feedback. We thank all the GitHub users who contributed code to scvi-tools over the years. We thank Nicholas Everetts for help with the analysis of the Drosophila data. We thank David Kelley and Nick Bernstein for help implementing Solo. We thank Marco Wagenstetter and Sergei Rybakov for help with the transition of the scGen package to use scvi-tools, as well as feedback on the scArches implementation. We thank Hector Roux de Bézieux for insightful discussions about the R ecosystem. We thank Kieran Campbell and Allen Zhang for clarifying aspects of the original CellAssign implementation. We thank the Pyro team, including Eli Bingham, Martin Jankowiak and Fritz Obermeyer, for help integrating Pyro in scvi-tools. Research reported in this manuscript was supported by the NIGMS of the National Institutes of Health under award number R35GM124916 and by the Chan-Zuckerberg Foundation Network under grant number 2019-02452. O.C. is supported by the EPSRC Centre for Doctoral Training in Modern Statistics and Statistical Machine Learning (EP/S023151/1, studentship 2420649). A.G. is supported by NIH Training Grant 5T32HG000047-19. A.S. and N.Y. are Chan Zuckerberg Biohub investigators.
Author information
Affiliations
Contributions
A.G., R.L and G.X. contributed equally. A.G. designed the scvi-tools application programming interface with input from G.X. and R.L. G.X. and A.G. led development of scvi-tools with input from R.L. G.X. reimplemented scVI, totalVI, AutoZI and scANVI with input from A.G. R.L. implemented Stereoscope with input from A.G. Data analysis in this manuscript was led by A.G., R.L. and G.X, with input from N.Y. A.G., R.L., P.B., E.M., M. Langevin., Y.L., J.S., G.M. and A.N., O.C. worked on the initial version of the codebase (scvi package), with input from M.I.J, J.R. and N.Y. R.L., E.M. and C.X. contributed the scANVI model, with input from J.R. and N.Y. A.G. implemented totalVI with input from A.S. and N.Y. T.A. implemented peakVI with input from A.G. A.G implemented scArches with input from M. Lotfollahi., F.J.T and N.Y. V.S. made several contributions to the codebase, including the LDVAE model. P.B. contributed the differential expression programming interface. E.d.V.B. and C.T.-L. provided tutorials on differential expression and deconvolution of spatial transcriptomics, with input from L.P. K.W. implemented CellAssign in the codebase with input from A.G. V.V.P.A., J.H. and M.J. made general code contributions and helped maintain scvi-tools. J.H. implemented LDA. T.A. and M.G. implemented MultiVI. V.K. improved Pyro support in scvi-tools and ported Cell2Location to use scvi-tools. N.Y. supervised all research. A.G., R.L., G.X., J.R. and N.Y. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
V.S. is a full-time employee of Serqet Therapeutics and has ownership interest in Serqet Therapeutics. F.J.T. reports consulting fees from Roche Diagnostics GmbH and Cellarity Inc., and ownership interest in Cellarity, Inc. N.Y. is an advisor to and/or has equity in Cellarity, Celsius Therapeutics and Rheos Medicines. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Martin Hemberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Supplementary information
Supplementary information
Supplementary Figs. 1–8, Notes 1–5, Tables 1–3 and References
Rights and permissions
About this article

Cite this article
Gayoso, A., Lopez, R., Xing, G. et al. A Python library for probabilistic analysis of single-cell omics data. Nat Biotechnol (2022). https://doi.org/10.1038/s41587-021-01206-w
https://www.nature.com/articles/s41587-021-01206-w
No hay comentarios:
Publicar un comentario