Debate Origen del Virus
Los autores del análisis indican que es muy poco probable que SARS-CoV-2 haya sido creado a partir de la manipulación de otro coronavirus. La estructura del genoma de SARS-CoV-2 no deriva de la de otro virus, que sería lo que se esperaría si fuera de diseño.
- https://genotipia.com/genetica_medica_news/sars-cov/
1-Qué información proporciona la secuenciación del genoma del coronavirus SARS-CoV-2





Amparo Tolosa, Genotipia
Conoce a tu enemigo. La conocida frase del famoso tratado de estrategia “El Arte de la guerra” adquiere un carácter esencial frente al nuevo agente microscópico que amenaza la salud de miles de personas y pone a prueba la organización y sistemas sanitarios de múltiples países: el coronavirus SARS-CoV-2.
Una de las mejores formas de conocer un organismo, es secuenciar su genoma, que contiene las instrucciones necesarias para hacerlo funcionar. Cuando se produce una pandemia como la de COVID-19, conocer el genoma del agente infeccioso responsable proporciona información con gran relevancia para los investigadores. Les permite identificar qué es lo que causa la enfermedad, conocer su origen y evolución con el tiempo o desarrollar estrategias terapéuticas para hacerle frente.
Identificación de SARS-CoV-2
La primera secuencia del genoma del agente infeccioso responsable de la epidemia iniciada en Wuhan se obtuvo en enero. Esta información fue crítica para identificar al virus como un coronavirus, muy similar al coronavirus responsable del Síndrome Agudo Respiratorio Grave (SARS en sus siglas en inglés), enfermedad respiratoria originada en Asia en 2003, que se propagó por diversos países. Los coronavirus son virus de ARN que se reciben su nombre por la característica estructura de su forma infectiva, similar a la corona solar. Este tipo de virus es responsable de muchos de los resfriados comunes, que no tienen consecuencias importantes sobre la salud. No obstante, otros pueden causar enfermedades mortales como el SARS o el síndrome respiratorio de oriente medio (MERS en sus siglas en inglés).
La similitud del nuevo coronavirus con el virus responsable del SARS, denominado SARS-CoV, fue determinante para denominar al nuevo virus SARS-CoV-2.

Características del nuevo coronavirus
El análisis del genoma de SARS-CoV-2, en combinación con las pruebas bioquímicas y las imágenes obtenidas por microscopía electrónica, permite conocer mejor sus características, incluyendo aquellas que pueden ser aprovechadas por los investigadores para desarrollar terapias. Así, a partir de pruebas bioquímicas y estructurales los investigadores han determinado que la parte más variable del genoma del coronavirus se encuentra precisamente en el dominio de unión al receptor de la proteína S, una proteína necesaria para la invasión del virus. En humanos, este dominio proteico tiene una afinidad especial por los receptores ACE2 de las células del hospedador.
Cuando los coronavirus infectan una célula, liberan en su interior su ARN que puede ser leído por la maquinaria celular para producir una larga cadena polipeptídica que es posteriormente fragmentada en péptidos funcionales para el virus. Otra característica encontrada en el genoma de SARS-CoV-2 es que esa cadena polipeptídica presenta un fragmento en que facilita la separación de los péptidos correspondientes a la proteína S.
Origen del Coronavirus de Wuhan
Una de las cuestiones más discutidas sobre el coronavirus SARS-CoV-2 es su origen. Las primeras investigaciones apuntaban a un posible origen animal, sin que estuviera claro cuál era exactamente. La hipótesis más aceptada en la actualidad es que el virus deriva de un virus de murciélagos que pasó a nuestra especie a través de un intermediario, como ocurrió en el caso de los coronavirus responsables del SARS y del MERS.
En paralelo a los estudios científicos, también empezaron a surgir también diversas teorías sobre si el coronavirus había sido creado en un laboratorio y liberado intencionada o accidentalmente. Un reciente análisis, que considera la información disponible del genoma del virus, descarta la creación del virus y plantea los posibles escenarios de su evolución.
Los autores del análisis indican que es muy poco probable que SARS-CoV-2 haya sido creado a partir de la manipulación de otro coronavirus. La estructura del genoma de SARS-CoV-2 no deriva de la de otro virus, que sería lo que se esperaría si fuera de diseño. Además, aunque el dominio proteico de unión al receptor de la proteína S mencionado anteriormente tiene afinidad por los receptores ACE2 humanos, esta afinidad no está optimizada mediante predicciones. Es diferente de lo que los algoritmos predictivos estimarían.
Los investigadores plantean diversos escenarios posibles de aparición del SARS-CoV-2. En el primero de ellos, el virus habría adquirido sus características a través de la selección natural en una especie animal antes de que el virus saltara a la especie humana. Entre los datos que apoyarían esta posibilidad está el hecho de que la variación del dominio de unión de la proteína S encontrada en SARS-CoV-2 es similar a la observada en otros coronavirus encontrados en pangolines. Sin embargo, los coronavirus más cercanos a SARS-CoV-2 presentes en animales no tienen la secuencia que favorece fragmentación de péptidos de la proteína S.
En el segundo escenario, la selección natural en el virus se habría producido en humanos tras la transferencia del virus desde una especie animal. La adquisición de la secuencia de corte habría sido incorporada también una vez en la especie humana.
Un tercer escenario posible planteado por los investigadores es que los cambios en el genoma de SARS-CoV-2 respecto a otros virus (es decir, su origen) hayan ocurrido por accidente en cultivos celulares de investigación. Los autores del análisis no encuentran evidencias de que ese haya sido el caso e indican que no creen que este escenario sea plausible. No obstante, señalan que con la información actual no se puede concluir de forma definitiva cuál de los tres escenarios es el correcto y será necesario investigar más.

Evolución del coronavirus y seguimiento de la enfermedad
La secuenciación de los genomas de los virus SARS-Cov-2 que se van aislando de las personas diagnosticadas como infectadas permite elaborar un registro de cómo va evolucionando el virus. Una característica de los virus de ARN como SARS-Cov-2 es que mutan rápidamente, y van acumulando cambios en su genoma. Registrar estos cambios es una herramienta epidemiológica muy útil para hacer el seguimiento de la infección.
Desde el inicio de la pandemia de COVID-19 los investigadores están compartiendo las secuencias de SARS-Cov-2 obtenidas en los diferentes países, lo que ha permitido elaborar un mapa dinámico de la evolución de SARS-Cov-2 que permite visualizar la progresión espacial y temporal del virus. Las primeras secuencias de los coronavirus aislados en España fueron incorporadas el pasado viernes, tras ser obtenidas por el Servicio de Secuenciación y Bioinformática de FISABIO y por el grupo de investigación en Epidemiología Molecular del I2SysBio, liderados por Fernando González-Candelas, catedrático de Genética de la Universitat de València.
Con la información proporcionada por los diferentes equipos, se sabe que el virus está mutando y que la tasa de mutación de SARS-Cov-2 es más lenta que la del virus de la gripe. Sin embargo, todavía no hay datos sobre si ha cambiado su virulencia. Conforme se obtengan más secuencias de más pacientes podrá mejorarse la resolución en el mapa de evolución del virus, y obtenerse más detalles.

Diseño de terapias
En la actualidad, el objetivo final de todas las investigaciones sobre el coronavirus SARS-Cov-2 es encontrar una forma de detenerlo y tratar a los pacientes. En este sentido conocer qué regiones del genoma del virus son más o menos susceptibles a tener mutaciones puede ser utilizado en el diseño de estrategias terapéuticas.
Por una parte, la similitud del genoma de SARS-Cov-2 con el del virus SARS-Cov-1 plantea la posibilidad de utilizar o adaptar estrategias que han mostrado potencial en SARS a la enfermedad causada por SARS-Cov-2. Por otra muchas de las aproximaciones para hacer frente a los virus dependen del reconocimiento de moléculas, bien ARN bien proteínas. En este sentido, interesará diseñar los agentes terapéuticos para que reconozcan moléculas con baja probabilidad de sufrir cambios.
En resumen, la secuenciación del genoma de SARS-Cov-2 es una herramienta esencial para estudiar la progresión y evolución del virus, así como para poder desarrollar tratamientos o vacunas para COVID-19.
Referencias:
Andersen KG, et al. The proximal origin of SARS-CoV-2. Nat Med. 2020. https://doi.org/10.1038/s41591-020-0820-9
Liu C, et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent Sci. 2020. Doi: https://doi.org/10.1021/acscentsci.0c00272
Fuentes:
Las primeras secuencias genómicas del virus SARS-CoV-2 de dos pacientes españoles se han obtenido en València. https://www.uv.es/instituto-biologia-integrativa-sistemas-i2sysbio/es/novedades-1285990801509/Novetat.html?id=1286122919661
¿De qué sirve secuenciar el coronavirus? https://theconversation.com/de-que-sirve-secuenciar-el-coronavirus-133717
¿Cómo cambian los virus? https://theconversation.com/como-cambian-los-virus-132996
https://genotipia.com/genetica_medica_news/sars-cov/
2-The proximal origin of SARS-CoV-2
Nature Medicine volume 26, pages 450–452 (2020)
To the Editor — Since the first reports of novel pneumonia (COVID-19) in Wuhan, Hubei province, China1,2, there has been considerable discussion on the origin of the causative virus, SARS-CoV-23 (also referred to as HCoV-19)4. Infections with SARS-CoV-2 are now widespread, and as of 11 March 2020, 121,564 cases have been confirmed in more than 110 countries, with 4,373 deaths5.
SARS-CoV-2 is the seventh coronavirus known to infect humans; SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease, whereas HKU1, NL63, OC43 and 229E are associated with mild symptoms6. Here we review what can be deduced about the origin of SARS-CoV-2 from comparative analysis of genomic data. We offer a perspective on the notable features of the SARS-CoV-2 genome and discuss scenarios by which they could have arisen. Our analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus.
Notable features of the SARS-CoV-2 genome
Our comparison of alpha- and betacoronaviruses identifies two notable genomic features of SARS-CoV-2: (i) on the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 appears to be optimized for binding to the human receptor ACE2; and (ii) the spike protein of SARS-CoV-2 has a functional polybasic (furin) cleavage site at the S1–S2 boundary through the insertion of 12 nucleotides8, which additionally led to the predicted acquisition of three O-linked glycans around the site.
1. Mutations in the receptor-binding domain of SARS-CoV-2
The receptor-binding domain (RBD) in the spike protein is the most variable part of the coronavirus genome1,2. Six RBD amino acids have been shown to be critical for binding to ACE2 receptors and for determining the host range of SARS-CoV-like viruses7. With coordinates based on SARS-CoV, they are Y442, L472, N479, D480, T487 and Y4911, which correspond to L455, F486, Q493, S494, N501 and Y505 in SARS-CoV-27. Five of these six residues differ between SARS-CoV-2 and SARS-CoV (Fig. 1a). On the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 seems to have an RBD that binds with high affinity to ACE2 from humans, ferrets, cats and other species with high receptor homology7.
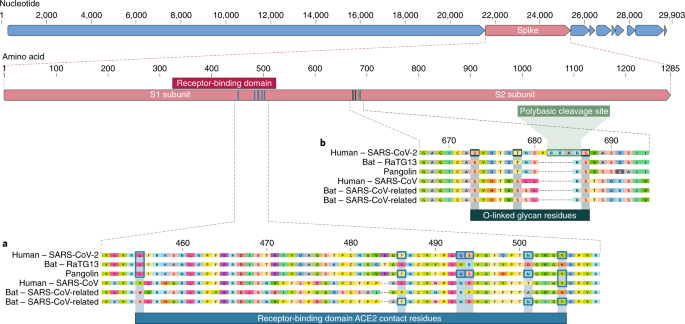
a, Mutations in contact residues of the SARS-CoV-2 spike protein. The spike protein of SARS-CoV-2 (red bar at top) was aligned against the most closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues in the spike protein that make contact to the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in lineage B betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146 and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378 (NCBI BioProject PRJNA573298)29,30.
While the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal7 and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding7,11. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation.
2. Polybasic furin cleavage site and O-linked glycans
The second notable feature of SARS-CoV-2 is a polybasic cleavage site (RRAR) at the junction of S1 and S2, the two subunits of the spike8 (Fig. 1b). This allows effective cleavage by furin and other proteases and has a role in determining viral infectivity and host range12. In addition, a leading proline is also inserted at this site in SARS-CoV-2; thus, the inserted sequence is PRRA (Fig. 1b). The turn created by the proline is predicted to result in the addition of O-linked glycans to S673, T678 and S686, which flank the cleavage site and are unique to SARS-CoV-2 (Fig. 1b). Polybasic cleavage sites have not been observed in related ‘lineage B’ betacoronaviruses, although other human betacoronaviruses, including HKU1 (lineage A), have those sites and predicted O-linked glycans13. Given the level of genetic variation in the spike, it is likely that SARS-CoV-2-like viruses with partial or full polybasic cleavage sites will be discovered in other species.
The functional consequence of the polybasic cleavage site in SARS-CoV-2 is unknown, and it will be important to determine its impact on transmissibility and pathogenesis in animal models. Experiments with SARS-CoV have shown that insertion of a furin cleavage site at the S1–S2 junction enhances cell–cell fusion without affecting viral entry14. In addition, efficient cleavage of the MERS-CoV spike enables MERS-like coronaviruses from bats to infect human cells15. In avian influenza viruses, rapid replication and transmission in highly dense chicken populations selects for the acquisition of polybasic cleavage sites in the hemagglutinin (HA) protein16, which serves a function similar to that of the coronavirus spike protein. Acquisition of polybasic cleavage sites in HA, by insertion or recombination, converts low-pathogenicity avian influenza viruses into highly pathogenic forms16. The acquisition of polybasic cleavage sites by HA has also been observed after repeated passage in cell culture or through animals17.
The function of the predicted O-linked glycans is unclear, but they could create a ‘mucin-like domain’ that shields epitopes or key residues on the SARS-CoV-2 spike protein18. Several viruses utilize mucin-like domains as glycan shields involved immunoevasion18. Although prediction of O-linked glycosylation is robust, experimental studies are needed to determine if these sites are used in SARS-CoV-2.
Theories of SARS-CoV-2 origins
It is improbable that SARS-CoV-2 emerged through laboratory manipulation of a related SARS-CoV-like coronavirus. As noted above, the RBD of SARS-CoV-2 is optimized for binding to human ACE2 with an efficient solution different from those previously predicted7,11. Furthermore, if genetic manipulation had been performed, one of the several reverse-genetic systems available for betacoronaviruses would probably have been used19. However, the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously used virus backbone20. Instead, we propose two scenarios that can plausibly explain the origin of SARS-CoV-2: (i) natural selection in an animal host before zoonotic transfer; and (ii) natural selection in humans following zoonotic transfer. We also discuss whether selection during passage could have given rise to SARS-CoV-2.
1. Natural selection in an animal host before zoonotic transfer
As many early cases of COVID-19 were linked to the Huanan market in Wuhan1,2, it is possible that an animal source was present at this location. Given the similarity of SARS-CoV-2 to bat SARS-CoV-like coronaviruses2, it is likely that bats serve as reservoir hosts for its progenitor. Although RaTG13, sampled from a Rhinolophus affinis bat1, is ~96% identical overall to SARS-CoV-2, its spike diverges in the RBD, which suggests that it may not bind efficiently to human ACE27 (Fig. 1a).
Malayan pangolins (Manis javanica) illegally imported into Guangdong province contain coronaviruses similar to SARS-CoV-221. Although the RaTG13 bat virus remains the closest to SARS-CoV-2 across the genome1, some pangolin coronaviruses exhibit strong similarity to SARS-CoV-2 in the RBD, including all six key RBD residues21 (Fig. 1). This clearly shows that the SARS-CoV-2 spike protein optimized for binding to human-like ACE2 is the result of natural selection.
Neither the bat betacoronaviruses nor the pangolin betacoronaviruses sampled thus far have polybasic cleavage sites. Although no animal coronavirus has been identified that is sufficiently similar to have served as the direct progenitor of SARS-CoV-2, the diversity of coronaviruses in bats and other species is massively undersampled. Mutations, insertions and deletions can occur near the S1–S2 junction of coronaviruses22, which shows that the polybasic cleavage site can arise by a natural evolutionary process. For a precursor virus to acquire both the polybasic cleavage site and mutations in the spike protein suitable for binding to human ACE2, an animal host would probably have to have a high population density (to allow natural selection to proceed efficiently) and an ACE2-encoding gene that is similar to the human ortholog.
2. Natural selection in humans following zoonotic transfer
It is possible that a progenitor of SARS-CoV-2 jumped into humans, acquiring the genomic features described above through adaptation during undetected human-to-human transmission. Once acquired, these adaptations would enable the pandemic to take off and produce a sufficiently large cluster of cases to trigger the surveillance system that detected it1,2.
All SARS-CoV-2 genomes sequenced so far have the genomic features described above and are thus derived from a common ancestor that had them too. The presence in pangolins of an RBD very similar to that of SARS-CoV-2 means that we can infer this was also probably in the virus that jumped to humans. This leaves the insertion of polybasic cleavage site to occur during human-to-human transmission.
Estimates of the timing of the most recent common ancestor of SARS-CoV-2 made with current sequence data point to emergence of the virus in late November 2019 to early December 201923, compatible with the earliest retrospectively confirmed cases24. Hence, this scenario presumes a period of unrecognized transmission in humans between the initial zoonotic event and the acquisition of the polybasic cleavage site. Sufficient opportunity could have arisen if there had been many prior zoonotic events that produced short chains of human-to-human transmission over an extended period. This is essentially the situation for MERS-CoV, for which all human cases are the result of repeated jumps of the virus from dromedary camels, producing single infections or short transmission chains that eventually resolve, with no adaptation to sustained transmission25.
Studies of banked human samples could provide information on whether such cryptic spread has occurred. Retrospective serological studies could also be informative, and a few such studies have been conducted showing low-level exposures to SARS-CoV-like coronaviruses in certain areas of China26. Critically, however, these studies could not have distinguished whether exposures were due to prior infections with SARS-CoV, SARS-CoV-2 or other SARS-CoV-like coronaviruses. Further serological studies should be conducted to determine the extent of prior human exposure to SARS-CoV-2.
3. Selection during passage
Basic research involving passage of bat SARS-CoV-like coronaviruses in cell culture and/or animal models has been ongoing for many years in biosafety level 2 laboratories across the world27, and there are documented instances of laboratory escapes of SARS-CoV28. We must therefore examine the possibility of an inadvertent laboratory release of SARS-CoV-2.
In theory, it is possible that SARS-CoV-2 acquired RBD mutations (Fig. 1a) during adaptation to passage in cell culture, as has been observed in studies of SARS-CoV11. The finding of SARS-CoV-like coronaviruses from pangolins with nearly identical RBDs, however, provides a much stronger and more parsimonious explanation of how SARS-CoV-2 acquired these via recombination or mutation19.
The acquisition of both the polybasic cleavage site and predicted O-linked glycans also argues against culture-based scenarios. New polybasic cleavage sites have been observed only after prolonged passage of low-pathogenicity avian influenza virus in vitro or in vivo17. Furthermore, a hypothetical generation of SARS-CoV-2 by cell culture or animal passage would have required prior isolation of a progenitor virus with very high genetic similarity, which has not been described. Subsequent generation of a polybasic cleavage site would have then required repeated passage in cell culture or animals with ACE2 receptors similar to those of humans, but such work has also not previously been described. Finally, the generation of the predicted O-linked glycans is also unlikely to have occurred due to cell-culture passage, as such features suggest the involvement of an immune system18.
Conclusions
In the midst of the global COVID-19 public-health emergency, it is reasonable to wonder why the origins of the pandemic matter. Detailed understanding of how an animal virus jumped species boundaries to infect humans so productively will help in the prevention of future zoonotic events. For example, if SARS-CoV-2 pre-adapted in another animal species, then there is the risk of future re-emergence events. In contrast, if the adaptive process occurred in humans, then even if repeated zoonotic transfers occur, they are unlikely to take off without the same series of mutations. In addition, identifying the closest viral relatives of SARS-CoV-2 circulating in animals will greatly assist studies of viral function. Indeed, the availability of the RaTG13 bat sequence helped reveal key RBD mutations and the polybasic cleavage site.
The genomic features described here may explain in part the infectiousness and transmissibility of SARS-CoV-2 in humans. Although the evidence shows that SARS-CoV-2 is not a purposefully manipulated virus, it is currently impossible to prove or disprove the other theories of its origin described here. However, since we observed all notable SARS-CoV-2 features, including the optimized RBD and polybasic cleavage site, in related coronaviruses in nature, we do not believe that any type of laboratory-based scenario is plausible.
More scientific data could swing the balance of evidence to favor one hypothesis over another. Obtaining related viral sequences from animal sources would be the most definitive way of revealing viral origins. For example, a future observation of an intermediate or fully formed polybasic cleavage site in a SARS-CoV-2-like virus from animals would lend even further support to the natural-selection hypotheses. It would also be helpful to obtain more genetic and functional data about SARS-CoV-2, including animal studies. The identification of a potential intermediate host of SARS-CoV-2, as well as sequencing of the virus from very early cases, would similarly be highly informative. Irrespective of the exact mechanisms by which SARS-CoV-2 originated via natural selection, the ongoing surveillance of pneumonia in humans and other animals is clearly of utmost importance.
References
- 1.
Zhou, P. et al. Nature https://doi.org/10.1038/s41586-020-2012-7 (2020).
- 2.
Wu, F. et al. Nature https://doi.org/10.1038/s41586-020-2008-3 (2020).
- 3.
Gorbalenya, A. E. et al. bioRxiv https://doi.org/10.1101/2020.02.07.937862 (2020).
- 4.
Jiang, S. et al. Lancet https://doi.org/10.1016/S0140-6736(20)30419-0 (2020).
- 5.
Dong, E., Du, H. & Gardner, L. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30120-1 (2020).
- 6.
Corman, V. M., Muth, D., Niemeyer, D. & Drosten, C. Adv. Virus Res. 100, 163–188 (2018).
- 7.
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. J. Virol. https://doi.org/10.1128/JVI.00127-20 (2020).
- 8.
Walls, A. C. et al. bioRxiv https://doi.org/10.1101/2020.02.19.956581 (2020).
- 9.
Wrapp, D. et al. Science https://doi.org/10.1126/science.abb2507 (2020).
- 10.
Letko, M., Marzi, A. & Munster, V. Nat. Microbiol. https://doi.org/10.1038/s41564-020-0688-y (2020).
- 11.
Sheahan, T. et al. J. Virol. 82, 2274–2285 (2008).
- 12.
Nao, N. et al. MBio 8, e02298-16 (2017).
- 13.
Chan, C.-M. et al. Exp. Biol. Med. 233, 1527–1536 (2008).
- 14.
Follis, K. E., York, J. & Nunberg, J. H. Virology 350, 358–369 (2006).
- 15.
Menachery, V. D. et al. J. Virol. https://doi.org/10.1128/JVI.01774-19 (2019).
- 16.
Alexander, D. J. & Brown, I. H. Rev. Sci. Tech. 28, 19–38 (2009).
- 17.
Ito, T. et al. J. Virol. 75, 4439–4443 (2001).
- 18.
Bagdonaite, I. & Wandall, H. H. Glycobiology 28, 443–467 (2018).
- 19.
Cui, J., Li, F. & Shi, Z.-L. Nat. Rev. Microbiol. 17, 181–192 (2019).
- 20.
Almazán, F. et al. Virus Res. 189, 262–270 (2014).
- 21.
Zhang, T., Wu, Q. & Zhang, Z. bioRxiv https://doi.org/10.1101/2020.02.19.950253 (2020).
- 22.
Yamada, Y. & Liu, D. X. J. Virol. 83, 8744–8758 (2009).
- 23.
Rambaut, A. Virological.org http://virological.org/t/356 (2020).
- 24.
Huang, C. et al. Lancet https://doi.org/10.1016/S0140-6736(20)30183-5 (2020).
- 25.
Dudas, G., Carvalho, L. M., Rambaut, A. & Bedford, T. eLife 7, e31257 (2018).
- 26.
Wang, N. et al. Virol. Sin. 33, 104–107 (2018).
- 27.
Ge, X.-Y. et al. Nature 503, 535–538 (2013).
- 28.
Lim, P. L. et al. N. Engl. J. Med. 350, 1740–1745 (2004).
- 29.
Wong, M. C., Javornik Cregeen, S. J., Ajami, N. J. & Petrosino, J. F. bioRxiv https://doi.org/10.1101/2020.02.07.939207 (2020).
- 30.
Liu, P., Chen, W. & Chen, J.-P. Viruses 11, 979 (2019).
Acknowledgements
We thank all those who have contributed sequences to the GISAID database (https://www.gisaid.org/) and analyses to Virological.org (http://virological.org/). We thank M. Farzan for discussions, and the Wellcome Trust for support. K.G.A. is a Pew Biomedical Scholar and is supported by NIH grant U19AI135995. A.R. is supported by the Wellcome Trust (Collaborators Award 206298/Z/17/Z―ARTIC network) and the European Research Council (grant agreement no. 725422―ReservoirDOCS). E.C.H. is supported by an ARC Australian Laureate Fellowship (FL170100022). R.F.G. is supported by NIH grants U19AI135995, U54 HG007480 and U19AI142790.
Author information
Affiliations
Corresponding author
https://www.ciencia.gob.es/portal/site/MICINN/menuitem.edc7f2029a2be27d7010721001432ea0/?vgnextoid=bb51a02169368710VgnVCM1000001d04140aRCRD
**El origen animal del SARS 2003 no se determinó hasta 14 años después de la epidemia. Y esperamos que un año después se determine y encuentre el origen del COVID?
3-La élite científica insiste en la fuga de laboratorio como posible origen de la pandemia
Un grupo de 18 de los científicos más importantes del mundo ha publicado una carta en 'Science' en la que aseguran que esta hipótesis no se puede descartar y que la conclusión de la OMS sobre el origen natural del SARS-CoV-2 no está científicamente justificada, y piden una nueva y exhaustiva investigación
Hace un año, la idea de que la pandemia de coronavirus (COVID-19) pudo haber sido causada por un accidente de laboratorio fue denunciada como teoría de la conspiración por parte de las principales revistas, científicos y organizaciones de noticias del mundo.
Pero, dado que el origen del virus que ya se ha llevado la vida de millones de personas sigue siendo un misterio, la posibilidad de que provenga de un laboratorio se ha convertido en una teoría que no se puede descartar.
En una carta publicada recientemente en la revista Science, 18 destacados biólogos, incluido el investigador de coronavirus más importante del mundo, ponen de manifiesto la necesidad de una nueva investigación de todos los posibles orígenes del virus y piden a los laboratorios y agencias de China que "abran sus registros" al análisis independiente. El texto afirma: "Debemos tomar en serio las hipótesis sobre el origen del contagio tanto de forma natural como de laboratorio hasta que tengamos suficientes datos".
La carta, coordinada por el microbiólogo de la Universidad de Stanford (EE. UU.) David Relman y el virólogo de la Universidad de Washington (EE. UU.) Jesse Bloom, hace referencia al reciente estudio sobre el origen de la COVID-19 realizado en conjunto por la Organización Mundial de la Salud (OMS) y China, que concluyó que probablemente un virus de murciélago llegó a los humanos a través de un animal intermedio y que un accidente de laboratorio fue "extremadamente improbable".
Pero, para los autores de la mencionada carta, esa conclusión no está científicamente justificada, ya que no se ha encontrado ningún rastro de cómo el virus habría pasado por primera vez a los seres humanos, y advierten que la posibilidad de un accidente de laboratorio solo ha sido estudiada de forma superficial. Solo un puñado de las 313 páginas del informe de la OMS sobre el origen del virus, junto con los anexos, se dedican a ese tema.
El conocido epidemiólogo de la Universidad de Harvard (EE. UU.) Marc Lipsitch, que se encuentra entre los firmantes de la carta, explica que no había expresado su opinión sobre el origen del virus hasta hace poco porque quería centrarse en mejorar el diseño de los estudios epidemiológicos y los ensayos de vacunas, en parte, porque el debate sobre la teoría del laboratorio se volvió muy controvertido. Y afirma: "Me mantuve al margen porque estaba ocupado lidiando con los efectos de la pandemia en vez del origen. [Pero] cuando la OMS publica un informe que hace una afirmación engañosa sobre un tema importante... hay que opinar sobre eso".
Varios de los firmantes de la carta, incluidos Lipsitch y Relman, ya habían reclamado un mayor escrutinio sobre la investigación de "ganancia de función", en la que los virus son modificados genéticamente para volverlos más contagiosos o virulentos. También se estaban realizando experimentos para diseñar patógenos en el Instituto de Virología de Wuhan (China), el principal centro de China para el estudio de virus de murciélagos similares al SARS-CoV-2. Algunos consideran el hecho de que la COVID-19 apareciera en la misma ciudad en la que se encuentra el laboratorio como evidencia circunstancial de que un accidente de laboratorio podría ser el culpable.
Lipsitch ya había estimado antes el riesgo de una pandemia causada por la fuga accidental de un laboratorio biológico de alta seguridad, con riesgos de 1 entre 1.000 y 1 entre 10.000 al año, y advirtió que la proliferación de miles de laboratorios de este tipo en todo el mundo era una gran preocupación.
Aunque los científicos chinos afirman que eso no es lo que pasó con el coronavirus, los redactores de la carta creen que es algo que solo se puede comprobar a través de una investigación más independiente. "Una investigación adecuada debe ser transparente, objetiva, basada en datos, que incluya una amplia experiencia, sujeta a supervisión independiente y gestionada de manera responsable para minimizar el impacto de los conflictos de intereses. Las agencias de salud pública y los laboratorios de investigación deben abrir sus registros al público. Los investigadores deberían documentar la veracidad y la procedencia de los datos a partir de los cuales se realizan los análisis y se extraen las conclusiones", afirma el texto.
La principal científica de enfermedades emergentes en el Instituto de Virología de Wuhan, Shi Zhengli, escribió en un correo electrónico que las sospechas de la carta estaban fuera de lugar y que dañarían la capacidad del mundo para responder a las pandemias. "Definitivamente es inaceptable. ¿Quién puede proporcionar una evidencia que no existe?", respondió Shi sobre la solicitud del grupo para ver los registros de su laboratorio.
"Es realmente triste leer esta 'Carta' escrita por estos 18 científicos prominentes. La hipótesis de una fuga de laboratorio se basa simplemente en la experiencia de un laboratorio que ha estado trabajando durante mucho tiempo en los coronavirus de murciélagos que están filogenéticamente relacionados con el SARS-CoV-2. Este tipo de afirmación definitivamente dañará la reputación y el entusiasmo de los científicos que se dedican a trabajar en los nuevos virus de animales que tienen un potencial riesgo de propagación a las poblaciones humanas y con el tiempo debilitará la capacidad de los humanos para prevenir la próxima pandemia", escribió Shi en su correo electrónico.
https://www.technologyreview.es/s/13389/la-elite-cientifica-insiste-en-la-fuga-de-laboratorio-como-posible-origen-de-la-pandemia?fbclid=IwAR3hwp8Gnl-G-HDjQ8XFpcPEFuu7-cllX4rX48CWBe5no23RFdtHQq6mn8o
4-Investigate the origins of COVID-19
See all authors and affiliations
- https://science.sciencemag.org/content/372/6543/694.1?fbclid=IwAR05jCSVgjNlwHKNz6NNvSivtOPaaRy6H22v5fkeirKAZQjTAvzVmp395gA
5-Bat cave study sheds new light on origin of SARS virus
Newly discovered SARS strains in bats hold genetic clues to the evolution of a human pandemic strain
- https://www.sciencedaily.com/releases/2017/11/171130141222.htm?fbclid=IwAR30D4X9qMYyfX2gpzDGzb8kcb7K5BtuHFIkOPSuVVUnWFb41H3wlW3bQ3Q
Como se señaló anteriormente, el RBD de SARS-CoV-2 se une de forma muy eficiente al ACE2 humano. Si se hubiera realizado la manipulación genética, probablemente se hubiera utilizado uno de los varios sistemas de genética reversa disponibles para los betacoronavirus. Los datos genéticos que se muestran en el estudio indican de forma irrefutable que el SARS-CoV-2 no deriva de ningún esqueleto de virus utilizado anteriormente.
En este mismo artículo sí proponen dos escenarios que pueden explicar con fundamento el origen del SARS-CoV-2:
- Selección natural en un hospedador animal antes de la transferencia zoonótica (del animal al ser humano).
- Selección
natural en humanos después de la transferencia zoonótica. También
discuten si un pase sucesivo de un coronavirus en cultivos celulares
humanos podría haber dado lugar al SARS-CoV-2. Esta hipótesis, aunque
parece muy improbable, no se puede descartar completamente por ahora.
- https://www.nationalgeographic.com.es/ciencia/coronavirus-ni-se-creo-ni-se-escapo-laboratorio_15452
- https://www.esciupfnews.com/2020/04/08/la-secuenciacion-del-coronavirus-sars-cov-2/
La investigación básica se centra en la bioquímica y las propiedades físicas que emplean los microbios que provocan enfermedades para dañar a su huésped. Dicha investigación considera también las características biofísicas de los microbios que se podrían usar en las vacunas o medicamentos útiles para prevenir o interrumpir el proceso de la enfermedad. A esta parte del ciclo de desarrollo de la vacuna se le conoce como investigación básica o preclínica
En el caso de la investigación para crear vacunas, los empleos como asistentes de investigación involucran hacer crecer líneas celulares en cultivo (una línea celular es un clon, o grupo de clones, que crecen en un cultivo, se derivan de una sola célula y pueden proliferar indefinidamente bajo condiciones estrictas de laboratorio), clonación de ADN o realización de ensayos (pruebas de laboratorio que buscan, cuantifican o miden alguna actividad de proteínas, virus y ADN). Los empleos en investigación básica ofrecen también oportunidades para convertirse en experto en el funcionamiento de equipo especializado de laboratorio, como citómetros de flujo que emplean rayos láser para evaluar células.
- https://ftp.historyofvaccines.org/index.php/es/contenido/articulos/carreras-profesionales-en-la-investigaci%C3%B3n-de-vacunas
6-Redfield se pueden infectar los investigadores
Redfield se refería al Instituto de Virología de Wuhan, el primer laboratorio de nivel 4 de bioseguridad de China.
Al señalar que “no es inusual que los patógenos respiratorios que se están trabajando en un laboratorio infecten al trabajador del laboratorio”, el exdirector explicó que “eso no implica ninguna intencionalidad". "Es mi opinión, ¿verdad? Pero soy virólogo. He pasado mi vida en virología”, afirmó.
El experto no cree que el virus que “se convirtió en uno de los más infecciosos que conocemos en la humanidad para la transmisión de persona a persona” haya pasado de un murciélago a un humano.
Redfield también afirmó que el virus ya se encontraba en circulación en septiembre u octubre de 2019.
A principios de febrero, la Organización Mundial de la Salud anunció que era “extremadamente improbable” que el virus se propagara desde un laboratorio en Wuhan.
Sin embargo, Peter Ben Embarek, líder del equipo de la OMS que realizó una investigación en Wuhan, aclaró que se necesita más trabajo para identificar la fuente de la pandemia.
Desde diciembre de 2019, cuando se detectó por primera vez el virus, se han reportado más de 125,6 millones de casos y más de 2,7 millones de muertes en todo el mundo, según un recuento de la Universidad Johns Hopkins.
- https://www.aa.com.tr/es/mundo/exdirector-de-autoridad-sanitaria-de-eeuu-afirma-que-el-coronavirus-naci%C3%B3-en-un-laboratorio-de-wuhan/2189551
7-Sobre el origen del SARS-CoV-2
Qué pasa, ¿qué ahora todos se han vuelto negacionistas y conspiranoicos?
El pasado 14 de mayo, un grupo de científicos de universidades como Harvard, Chicago, Toronto, Cambridge, Yale, Stanford, Berkeley y del California Institute of Technology y Massachusetts Institute of Technology, publicaron una carta en la revista Science que la que solicitaban seguir estudiando sobre el origen del SARS-CoV-2: “Debemos tomar en serio las hipótesis sobre el origen tanto natural como de laboratorio hasta que tengamos suficientes datos. La investigación debe ser transparente, objetiva, basada en datos, que incluya una amplia experiencia, que esté sujeta a una supervisión independiente y que se gestione de manera responsable para minimizar el impacto de los conflictos de intereses. Las agencias de salud pública y los laboratorios de investigación deben abrir sus registros al público. Los investigadores deben documentar la veracidad y la procedencia de los datos a partir de los cuales se realizan los análisis y se extraen las conclusiones, de modo que los análisis sean reproducibles por expertos independientes”.
Qué pasa, ¿qué ahora todos se han vuelto negacionistas y conspiranoicos?
- Si me preguntas, ¿es posible hoy en día crear un nuevo virus artificial en el laboratorio?
La respuesta es, si (mira por ejemplo Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice).
- ¿Es posible que un virus se escape de un laboratorio de seguridad?
La respuesta es, si (mira por ejemplo Laboratory-acquired severe acute respiratory síndrome o Brucellosis confirmed in 65 people from Lanzhou veterinary institute o El descubrimiento del virus Marbug).
Entonces, ¿el SARS-CoV-2 se ha creado artificialmente y se ha escapado de un laboratorio? No podemos descartarlo, pero lo más probable es que no. Una cosa es que sea posible, otra distinta qué es lo más probable.
Los coronavirus son virus de animales
Los coronavirus (CoV) de animales se conocen desde finales de los años 30 (del siglo pasado). Son una gran familia dentro de los virus, compuesta por cuatro géneros (alfa, beta, gamma y delta-CoV). El género beta-CoV contiene a la mayoría de los que infectan a humanos y se subdivide a su vez en cuatro linajes (A, B, C y D). El origen de la mayoría de los alfa y beta-CoV está en los murciélagos y roedores, mientras que las aves son el mayor reservorio natural de los gamma y delta-CoV. Desde hace miles de años, los CoV han estado constantemente cruzando la barrera entre especies.

Una de las características de los coronavirus es su tremenda capacidad de saltar de una especie animal a otra
Los CoV causan, principalmente, enfermedades respiratorias y gastrointestinales en muchos animales de granja y domésticos: el virus de la bronquitis infecciosa de las aves, el coronavirus respiratorio canino, la hepatitis murina, el coronavirus bovino, el virus de la gastroenteritis transmisible en cerdos, la peritonitis infecciosa felina y un largo etcétera. Los CoV se aíslan de aves, ratones, ganado vacuno, cerdos, gatos, perros, animales silvestres, …. Uno de los animales que más tipos diferentes de coronavirus alberga y que, por tanto, actúa como un almacén o reservorio natural de este tipo de virus son … los murciélagos. Se han identificado más de 200 tipos distintos de CoV en los murciélagos y el 35% del viroma (el conjunto de genomas de virus) del murciélago son CoV. (Por cierto, los murciélagos no son roedores, son los únicos mamíferos voladores, de los que existen más de 1.200 especies distintas, representan aproximadamente un 20% de todas las especies de mamíferos, y están presentes en todos los continentes, excepto en la Antártida. Algunas de sus colonias pueden albergar cientos de miles de individuos).
Los coronavirus humanos
En 1965 se describieron un nuevo tipo de virus respiratorios humanos, “parecidos al virus de la gripe”, muy difíciles de cultivar en el laboratorio y que solo se podían detectar infectando voluntarios. La naturaleza exacta de esos virus era un misterio, hasta que en 1967, una mujer, June Almeida, desarrolló un nuevo método para poder verlos por microscopía electrónica. La técnica, absolutamente novedosa, consistía en emplear anticuerpos marcados que se unían a las partículas virales y así poderlas ver al microscopio. Las imágenes que los investigadores obtuvieron les recordaban al halo que se observa en el sol, la corona solar y decidieron llamarlos corona-virus. Habían nacido un nuevo tipo de virus respiratorios: los coronavirus humanos (HCoV).
En humanos, además del SARS-CoV-2, se conocen otros seis coronavirus que causan enfermedad. Cuatro de ellos causan una infección leve y se denominan seguido de un código: HCoV-229E, HCoV-OC43, HCoV-NL63 y HCoV-HKU1. El 30% de los catarros comunes están producidos por estos cuatro coronavirus, en algunos casos también cursan con trastornos digestivos, y en niños y personas mayores inmunocomprometidas pueden llegar a ser graves. Su distribución es global y tienden a ser estacionales (invierno en climas templados). El primero que se descubrió fue el HCoV-229E, que se aisló del tracto respiratorio de un paciente en 1966. Al año siguiente, se aisló el HCoV-OC43. A finales de 2004 se descubrió un nuevo CoV, HCoV-NL63 aislado de un bebé de siete meses en Holanda. Se ha encontrado sobre todo en niños pequeños, ancianos y personas inmunocomprometidas. El mismo año se aisló el HCoV-HKU1 de un paciente de 71 años hospitalizado por neumonía y bronquiolitis en Hong Kong.
En 2002, se describió el SARS-CoV (8.096 casos y 774 muertos) que causaba una neumonía aguda y severa. Se extendió por 27 países. Diez años después, en 2012 apareció el MERS-CoV, un nuevo coronavirus que causó el síndrome respiratorio de Oriente Medio, y que aunque infectó a menos personas, unas 2.500, su letalidad fue mucho mayor, de hasta el 35%. El MERS-CoV todavía se aísla de forma esporádica.
Todos estos HCoV tiene un origen animal. HCoV-NL63, HCoV-229E, SARS-CoV y MERS-CoV se han originado en CoV de murciélagos, mientras que el origen de HCoV-OC43 y HCoV-HKU1 está en los roedores.

A este tipo de virus que infectan al ser humano pero que tienen un origen animal se les denomina zoonóticos. Más del 70% de los patógenos humanos emergentes son de origen animal.
En este salto desde el murciélago o el roedor al ser humano ha habido otros animales que han actuado como intermediarios y donde los virus se han ido adaptado para infectar al ser humano: las civetas y los mapaches en el caso del SARS-CoV, los dromedarios en MERS-CoV, o el ganado vacuno en HCoV-OC43. Murciélagos y roedores actuarían, por tanto, como reservorio natural o lugar donde los ancestros de estos HCoV viven y se multiplican y son el origen común de infecciones en otros animales. En los hospedadores intermedios, los CoV se irían adaptado al ser humano.
Actualmente, los HCoV-229E, HCoV-OC43, HCoV-NL63 y HCoV-HKU1 están muy adaptados al ser humano, son fácilmente transmisible y causan infecciones leves. Por el contrario, SARS-CoV y MERS-CoV son mucho más patógenos, no están tan bien adaptados y por eso su transmisión entre humanos no es tan frecuente (desde 2004 no se ha vuelto a detectar ningún caso de SARS-CoV, y los brotes de MERS-CoV siguen estando asociados al contacto con dromedarios que actúan como reservorio intermedio del virus).
La familia de coronavirus es muy diversa, se mezclan entre ellos y saltan de una especie animal a otra. No podemos descartar por tanto, que otro nuevo coronavirus como SARS-CoV y MERS-CoV vuelva a aparecer y a darnos problemas.
El origen de SARS-CoV-2
Una de las zonas del genoma más interesantes del SARS-CoV-2 para investigar su origen es la que codifica para la proteína S, porque es la más variables y porque su función es esencial para la entrada en la célula. La proteína S (de spike) forma esas espículas que se proyectan hacia al exterior y que le dan el nombre al corona-virus. El SARS-CoV-2 inicia la entrada en las células humanas después de que la proteína S se una al receptor de la membrana celular, que en este caso es el ACE2. La función biológica de este receptor ACE2 es la maduración de la angiotensina, una hormona que controla la vasoconstricción y la presión arterial. ACE2 es una proteína de membrana que se expresa en pulmones, el corazón, los riñones y el intestino.
La proteína S es la llave de entrada del virus a la célula y la cerradura en la célula es el receptor ACE2. Los modelos en 3D demuestran que en este proceso, la proteína S se divide en dos subunidades, S1 y S2, que se separan por la acción de una enzima de la célula con actividad proteasa, que se denomina furina. Así, S1 se une a su receptor ACE2 y el otro fragmento S2 es escindido a su vez por otra proteasa de la superficie de la célula humana, denominada TMPRSS2. Como resultado la envoltura de virus se fusiona con la membrana de la célula y el virus entra en su interior. Por tanto, la subunidad S1 se encarga de la unión al receptor, mientras que S2 es responsable de la fusión de las membranas.

Modelo en 3D de la proteína S de SARS-CoV-2 (Fuente).
Los análisis estructurales, genómicos y bioquímicos de esa proteína S nos permiten estudiar este proceso en detalle y demuestran que SARS-CoV-2 posee dos particularidades importantes, que pueden relacionarse con su origen.
1. El dominio RBD de la proteína S tiene una alta afinidad por el receptor ACE2
En primer lugar, la proteína S de SARS-CoV-2 posee una secuencia que se denomina RBD (dominio de unión al receptor), la parte más variable del genoma del virus, en la que hay seis aminoácidos que son esenciales para unirse al receptor ACE2. Si comparamos esa secuencia entre SARS-CoV-2 y el SARS-CoV, solo un aminoácidos de esos seis es común. La proteína S de SARS-CoV-2 tiene, por tanto, un dominio RBD que se une con una muy alta afinidad al receptor ACE2 de humanos, pero también de otras especies animales con una alta homología en ese receptor, como hurones o gatos. Esta alta afinidad por el receptor ACE2 probablemente influye en la alta capacidad de infectar las células que tiene este virus. Sin embargo, los análisis computacionales indican que ese dominio no es el mejor posible para unirse al receptor, teóricamente puede haber otras combinaciones que sean aún más eficaces para unirse al receptor. Esto sugiere que esa secuencia ha surgido por un proceso de selección natural a lo largo de pases del virus entre personas o animales. Si fuera un producto manipulado por ingeniería genética, lo habríamos hecho mejor. Si alguien hubiera diseñado este nuevo virus para que fuera patógeno lo hubiera hecho mejor.
2. La proteína S posee una secuencia de corte por furina
La otra particularidad de la proteína S de SARS-CoV-2 tiene que ver con el sitio de unión entre esas dos subunidades, S1 y S2, de las que está formada. En SARS-CoV-2 esa proteína S tiene una secuencia entre esas subunidades que permite el corte por la enzima de la célula, la furina, y por otras proteasas. Eso determina la infectividad del virus y su rango de hospedador, a qué células o animales puede infectar. Aunque algunos HCoV, como el HKU1, también tienen esa característica, el sitio de corte por furina no es muy frecuente en todos los coronavirus, y menos en los del grupo beta, al que pertenece el SARS-CoV-2. Esta secuencia tan peculiar, ¿podría ser fruto de la manipulación genética del virus? Si lo comparamos con lo que ocurre en el virus de la gripe, muy probablemente se haya generado también por selección natural.

Características de la proteína S de SARS-CoV-2 y otros coronavirus relacionados. Se detalla de forma progresiva la secuencia de nucleótidos del genoma, la secuencia de aminoácidos de la proteína S con sus dos subunidades S1 y S2, el dominio de unión al receptor (RBD) y la zona de corte por furina (polybasic clevage site). (Fuente)
En algunos virus de la gripe aviar se ha visto que en situaciones de alta densidad de poblaciones de aves, se selecciona de forma natural este tipo de secuencias de corte en la hemaglutinina de la envoltura (similar a la proteína S del coronavirus). Esto hace que el virus se replique más rápidamente y sea más transmisible. Así es cómo algunos virus de gripe aviar de baja patogenicidad se convierten en virus de alta patogenicidad. También se ha observado la adquisición de estos sitios de corte en la hemaglutinina después de pases repetidos del virus en cultivo celular o en animales. Por lo tanto, esta nueva propiedad es fruto de la selección natural. Lo mismo ha podido ocurrir en el coronavirus.
Si el origen del genoma de SARS-CoV-2 fuera la ingeniería genética, muy probablemente se habrían empleado algunos sistemas genéticos ya presentes en otros beta-CoV y los datos no demuestran nada de esto. Por el contrario, lo más probable es que estas dos características del virus sean fruto de la selección natural y para ello hay dos posibles escenarios: que se haya seleccionado en un animal antes de transferirse al ser humano; o que la selección haya ocurrido en el ser humano después de su transferencia desde un animal.
3. Selección en un animal antes de transferirse a humanos
Desde el inicio, el origen de SARS-CoV-2 se ha relacionado con el mercado de animales vivos de Wuhan. Cuando se comparan los genomas de los CoV, el más parecido al SARS-CoV-2 es el aislado de un murciélago en Yunnan (China) en 2013, el genoma RaTG13 de Rhinolophus affinis, con más de un 96% de identidad. Sin embargo, cuando se compara la zona RBD de la proteína S difieren significativamente. En otros estudios, se han analizado muestras de varios pangolines (Manis javanica) que llegaron a China por contrabando entre 2017 y 2018, y han detectado coronavirus con una similitud entre el 85 y el 92% con el SARS-CoV-2. Aunque el virus del murciélago sigue teniendo una homología a nivel del genoma mayor, la similitud entre el SARS-CoV-2 y los coronavirus del pangolín era especialmente alta en el dominio RBD de la glicoproteína S, incluidos los seis aminoácidos característicos de esa zona en SARS-CoV-2. Esto refuerza la idea de que la optimización de la proteína S para unirse al receptor ACE2 humano es fruto de la selección natural y no de ingeniería genética o de pases sucesivos del virus en un laboratorio.
Sin embargo, ni los coronavirus de murciélagos ni los de los pangolines tienen el sitio de corte de furina en la proteína S. Los coronavirus son muy frecuentes entre estos y otros animales y es muy probable que todavía no hayamos dado con el precursor animal del SARS-CoV-2. No podemos descartar que fenómenos de mutación, inserción y deleción hayan ocurrido de forma natural en el gen S en algún otro animal, probablemente con alta densidad de población y con un receptor ACE2 similar al humano.
4. Selección en humanos después de su transferencia desde un animal
Otra posibilidad es que el SARS-CoV-2 haya adquirido esas características mientras se transmitía de forma indetectable entre humanos. Todos los genomas de SARS-CoV-2 secuenciado hasta ahora demuestra que tienen un origen clonal a partir de un ancestro común en Wuhan, muy probablemente a principios de noviembre de 2019. La presencia en los pangolines del mismo dominio RBD en la proteína S sugiere que esa característica ya estaba en el virus antes de su salto a humanos. Quizá, entonces, el sitio de corte por furina fue el que se seleccionó durante la transmisión entre humanos. Esto presupone que el virus estaba presente antes de noviembre de 2019 y que se transmitía entre nosotros de forma indetectable durante un tiempo. Eso ahora no lo sabemos, pero sería muy interesante si somos capaces de hacer estudios retrospectivos y comprobar si realmente el virus circulaba entre nosotros antes de su estallido en Wuhan a finales de 2019.
El hecho de que SARS-CoV-2 entró en los seres humanos a partir de un origen animal implica que la probabilidad de futuros brotes es muy alta, ya que virus similares siguen circulando en la población animal y podrían volver a saltar a los seres humanos.
Como vemos las peculiares características de SARS-CoV-2 ya estaban en la naturaleza y no hay que imaginar experimentos de laboratorio para explicar su origen. Conocemos menos del 1% de los virus que hay ahí fuera y más del 70% de los nuevos virus emergentes tienen su origen en los animales. Los virus son millones de millones de “individuos”, que se multiplican a una velocidad enorme y con una frecuencia de mutación y recombinación extraordinaria. Los virus no es que muten, es que viven mutando. En ellos, la evolución va a cámara rápida. La naturaleza tiene suficientes recursos como para generar este y otros muchísimos virus.
5. SARS-CoV-2 en animales
Otro dato interesante es que hay una gran cantidad de animales que o son susceptibles a una infección experimental con SARS-CoV-2 o lo han adquirido de forma natural. Entre los que se han podido infectar experimentalmente están: gatos domésticos, perros, hurones, visones, hámster, algunas especies de ratas y ratones, macacos, mono verde africano, musarañas, murciélagos frugívoros, mapaches, conejos de laboratorio, ganado bovino, …. Por el contrario, no se ha conseguido infección experimental en ardillas, perros de la pradera, ratones domésticos y un tipo de murciélagos gigantes marrones. Los estudios con ratones de laboratorio mostraron que aunque no eran susceptibles a una infección experimental con la cepa ancestral de SARS-CoV-2, dos variantes que han surgido en humanos sí que dieron lugar a la replicación del virus en los pulmones. Este es un hallazgo importante, ya que demuestra que el rango de hospedadores de SARS-CoV-2 puede ampliarse conforme el virus vaya evolucionando y vayan surgiendo nuevas variantes.
Por otra parte, se han demostrado infecciones adquiridas naturalmente de SARS-CoV-2 en perros, gatos y hurones en entornos domésticos, en tigres, leones, pumas y leopardos en colecciones zoológicas, en gorilas y en granjas de visón americano. De momento, las granjas de visones son la única evidencia de mantenimiento de una infección adquirida naturalmente en una población animal y salto a los humanos.
Lo más probable es un origen natural, pero ¿es posible otra hipótesis?
A pesar de todo lo que acabamos de decir, es verdad que hay dudas razonables sobre qué se hacía y cómo se trabajaba en el Instituto de Virología de Wuhan.
China tardó ¡un año! (14 de enero de 2021) en permitir que un equipo internacional de la OMS visitara Wuhan para investigar sobre el origen del virus. Su conclusión fue que "muy probable" el SARS-CoV-2 tuviera un origen animal, aunque no se sabe cuál. Desgraciadamente, fue el Gobierno chino el que recogió los datos y las muestras y recopiló toda la información, mientras que el equipo internacional solo pudo trabajar sobre esos datos e informes.
Por otra parte, se sabía que desde antes de 2008, se venían realizando experimentos de manipulaciones genéticas de los coronavirus SARS y MERS, denominadas “ganancias de función”, para mejorar su capacidad de infección y transmisión.
Desde 2014, el Gobierno estadounidense había establecido una moratoria a la financiación de este tipo de experimentos por su peligrosidad y un potencial pandémico.
En marzo de 2020, los máximos responsables del Instituto señalaron que ningún trabajador del mismo había dado positivo en los tests de detección del SARS-CoV-2. Pero, recientemente se ha hecho público que al menos tres científicos del Instituto enfermaron con síntomas compatibles de COVID-19 un mes antes del anuncio oficial de la existencia de un nuevo coronavirus, por lo que sigue habiendo serias dudas sobre el nivel de bioseguridad del Instituto. En un informe de 2018 de técnicos del Departamento de Estado de EE.UU. para verificar la bioseguridad de las instalaciones del Instituto, se mostraba la preocupación por la falta de seguridad, debilidades de gestión del laboratorio y falta de personal especializado, y describía que muchos de los trabajos no se hacían dentro de las instalaciones de BSL4.
CONCLUSIÓN: con los datos que tenemos en este momento, la hipótesis más probable es que el SARS-CoV-2, como el resto de CoV humanos, sea de origen natural, a partir de un reservorio natural de CoV de murciélagos y a través de alguna especie intermedia (todavía sin identificar) donde se fue adaptando al ser humano. La naturaleza tiene suficiente recursos para generar este y cualquier otro virus. Sin embargo, la tremenda opacidad y falta de trasparencia del Gobierno Chino hace que no se pueda descartar como hipótesis, menos probable pero posible, un origen en el laboratorio. Solo una investigación transparente, objetiva, basada en datos, e independiente nos dirá la verdad.
Referencias:
- The proximal origin of SARS-CoV-2. Andersen, K.G., et al. Nat Med (2020). https://doi.org/10.1038/s41591-020-0820-9
- Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Renhong Y., et al. Science 27 Mar 2020. Vol. 367, Issue 6485, pp. 1444-1448.
- Zoonotic origins of human coronaviruses. Ye, Z-W, y col. Review Int J Biol Sci. 2020 Mar 15;16(10):1686-1697. doi: 10.7150/ijbs.45472.
- Zoonotic origins and animal hosts of coronaviruses causing human disease pandemics: A review. Latif, AA, y col. Review Onderstepoort J Vet Res. 2020 Dec 21;87(1):e1-e9. doi: 10.4102/ojvr.v87i1.1895.
- Natural selection versus creation: a review on the origin of SARS-COV-2. Barh, D., y col. Review Infez Med. 2020 Sep 1;28(3):302-311.
- TWiV 762: SARS-CoV-2 origins with Robert Garry (May 30, 2021).
https://microbioun.blogspot.com/2021/05/sobre-el-origen-del-sars-cov-2.html
8.¿Cuál es el origen del coronavirus?
En la comunidad científica se respira indignación por la opacidad generalizada de la actividad científica asiática. Pocos lo han manifestado públicamente durante la pandemia para no contribuir a la desinformación y al miedo.


Conocer el origen del coronavirus es fundamental para mitigar el riesgo de brotes futuros. Esta pregunta ha recuperado protagonismo en los medios por una carta publicada el 14 de mayo en la revista Science en la que un grupo de científicos solicita una investigación más profunda, objetiva y transparente sobre el origen del SARS-CoV-2.
Se está hablando de tres opciones: un proceso natural de zoonosis (enfermedad transmitida de animales a humanos), un fallo de seguridad en un laboratorio en el que se produjo la liberación accidental del virus, y una creación deliberada de un virus con potencial pandémico. Esta última opción es un disparate, como veremos a continuación. Pero las otras dos son hipótesis posibles, lo que no implica que ambas sean igual de probables. De hecho, la balanza de la evidencia científica se inclina hacia un proceso de selección natural.
Existen cientos de coronavirus diferentes. Desde hace miles de años han estado cruzando las barreras entre especies. Principalmente afectan a animales mamíferos. El murciélago es un gran reservorio de coronavirus. Siete de ellos afectan a humanos, la mayoría solo causan catarros y gastroenteritis leves. El origen de todos ellos se explica por procesos de zoonosis a partir de virus de murciélagos y roedores, y a través de intermediarios como los mapaches o los pangolines en los que se irían adaptando evolutivamente hasta infectar a humanos.
Este proceso ocurre realmente rápido: los virus se reproducen a gran velocidad dentro de las células, mutando con celeridad, lo que hace que la evolución en ellos vaya a cámara rápida. Por eso solo conocemos un 1% de los virus que existen, y más del 70% tienen origen animal conocido.
La mayoría de los coronavirus se contagian con facilidad y producen enfermedades leves. En cambio, el primer SARS y el MERS son más patógenos pero se contagian con dificultad entre humanos, por eso los casos se pueden contener con cierta facilidad. Sin embargo, el SARS-CoV-2 tiene una llave que se adapta mejor a la cerradura de entrada en la célula, lo que propicia la transmisión. Esta llave es la proteína S que encaja con la cerradura ACE2 de las células humanas.
Se ha especulado mucho acerca de si la proteína S tenía un origen sintético, pero la realidad es que los estudios en ingeniería genética revelan que el proceso de llave-cerradura aunque es funcional, no está optimizado: la proteína S se tiene que dividir en dos subunidades por mediación de una enzima proteasa a través de un corte por furina propiciando que la envoltura del virus se fusione con la membrana celular y así introducir el material genético del virus en la célula. Es decir, hay varios pasos en este proceso que en ingeniería genética serían considerados una chapuza, que por manipulación artificial se habrían resuelto mucho mejor y de un modo muy diferente.
Además, este tipo de proceso llave-cerradura es muy similar al de los coronavirus que afectan a los pangolines, que son un hospedador intermedio, y el "corte por furina" pudo haberse seleccionado naturalmente una vez comenzó la transmisión entre humanos. Así que todo apunta a que el SARS-CoV-2 es fruto de la selección natural.
Ahora se cumple un año desde que la Organización Mundial de la Salud, en colaboración con las autoridades chinas, emprendieron un estudio sobre el origen del coronavirus cuyos resultados se publicaron en noviembre de 2020. Se barajaron las dos hipótesis: un accidente de laboratorio o un proceso natural de zoonosis. Los datos fueron aportados por las autoridades chinas. A partir de ellos se llegó a la conclusión de que la zoonosis es "muy probable" y el accidente "extremadamente improbable". No obstante, las dos posibilidades no recibieron una consideración equilibrada, por lo que la OMS, las autoridades estadounidenses y europeas, entre otros, manifiestan que hay lagunas en la investigación y solicitan acceso directo y libre a las muestras, los datos y las instalaciones chinas de interés.
En el pasado se produjeron contagios accidentales con virus consecuencia de fallos de seguridad en el laboratorio. Ocurrió con la viruela, con la brucelosis, con la fiebre hemorrágica o con el ébola. Es cierto que fueron casos rotundamente excepcionales, pero el riesgo de cometer un error nunca es cero, ni para el trabajo científico ni para cualquier otro trabajo. Por eso también se investiga esta hipótesis porque, aunque es improbable, sí es posible.
En la comunidad científica se respira indignación por la opacidad generalizada de la actividad científica asiática. Pocos lo han manifestado públicamente durante la pandemia para no contribuir a la desinformación y al miedo. Sin embargo, al comienzo fueron algunos médicos, científicos, periodistas y ciudadanos chinos quienes compartieron con el mundo a título individual información crucial sobre la propagación del virus, a menudo a un gran costo personal y profesional.
Recientemente se hizo público que al menos tres científicos del Instituto de Virología de Wuhan (China) enfermaron con síntomas compatibles con la COVID-19 un mes antes del anuncio oficial de un nuevo coronavirus, por lo que hay serias dudas acerca del nivel de bioseguridad del Instituto de Wuhan. Ya en 2018, en un informe para verificar la bioseguridad de las instalaciones, los técnicos del Departamento de Estado de los EEUU expresaron su preocupación por la falta de seguridad, las debilidades en la gestión del laboratorio y la falta de personal especializado, y describieron que muchos de los trabajos no se estaban realizando con el nivel 3 o 4 de biocontención necesario.
Los ensayos con patógenos potencialmente pandémicos se realizan en instalaciones de biocontención de al menos nivel 3 (BSL3). Para exhibir la marca BSL3, un laboratorio nunca debe intercambiar aire o material no estéril con el exterior. Para ello, toda la instalación tiene presión negativa y siempre, todos los días y noches del año, todo el aire pasa por filtros HEPA. El sistema de entrada y salida de personas y equipos cuenta con duchas y puertas dobles conmutadas que nunca se abren al mismo tiempo, ni sin esterilizar los residuos y descontaminar al personal.
Para estudiar cómo un virus puede volverse potencialmente pandémico se pueden inducir cambios genéticos deliberados. A este tipo de estudios se les denomina de "ganancia de función". Estos estudios generan controversia por su potencial peligrosidad. Por un lado, las consecuencias de un accidente podrían ser catastróficas. Por otro lado, gracias a ellos se consiguió la vacuna para el sarampión o detectar y detener brotes de gripe aviar.
También hay quien se opone a estos estudios por temor a que alguien use estos virus modificados como arma biológica. Es una idea perezosa, puesto que prácticamente cualquier avance científico y tecnológico podría utilizarse con fines malvados.
En EEUU y en Europa hay una moratoria de financiación desde 2014 que impide realizar estudios de ganancia de función en virus primos del SARS-CoV-2, como el MERS y el SARS-CoV, por su potencial riesgo pandémico. No obstante, en China se llevan haciendo desde 2007, así que la hipótesis de un contagio accidental con un virus experimental es posible. Por eso, aunque lo más probable sea el origen natural del virus, esta otra hipótesis, aunque muy improbable, es posible, y merece que se investigue en profundidad.
La investigación sobre el origen del coronavirus no pretende señalar culpables, ni promover la hostilidad intercultural, sino garantizar la independencia y trasparencia propia de la actividad científica. Saber cómo surgió la COVID-19 es fundamental para mitigar el riesgo de brotes futuros.
Si el origen del coronavirus fuese un accidente de laboratorio, la forma de evitar que esto no se repita es obvia, no por ello fácil. Sin embargo, si el origen del coronavirus es la selección natural, como todo parece indicar, no me produce ningún alivio. Al contrario. Hay muchos virus similares al SARS-CoV-2 circulando entre animales que podrían volver a saltar a los humanos, conque el origen natural del coronavirus implica que la probabilidad de futuros brotes es muy alta. Así que sea cual sea el origen del coronavirus, ninguna de las respuestas es despreocupada.
https://www.lasexta.com/el-muro/deborah-garcia/cual-origen-coronavirus_2021060260b730894e67580001cb88c5.html?fbclid=IwAR1metx9WCM5m9AG8gSwiTZuM5jKC1T4mQY1GglYI-7Efr9NAlL-I-qmeTM9 . Un breve hilo sobre las filtraciones virales de laboratorio a lo largo de la historia:
- https://twitter.com/chris_said/status/1396827966395625474?s=21&fbclid=IwAR0RTZktyOoDdqx_IFWGQGoIBD4a8k3L74WdgRNO_d3ZFkzTBpJsfmNLPCU
Threatened pandemics and laboratory escapes: Self-fulfilling prophecies
El SARS-1 se ha escapado de sofisticados laboratorios en 6 ocasiones distintas, en Singapur, Taiwán y China.- https://thebulletin.org/2014/03/threatened-pandemics-and-laboratory-escapes-self-fulfilling-prophecies/
Smallpox escaped 3 times from labs in England, causing 80 cases and 3 deaths.
https://t.co/iWgpCelOBf?amp=1
The 2007 Foot and mouth disease outbreak in the UK first appeared four kilometers from a biosafety level 4 laboratory
https://en.wikipedia.org/wiki/2007_United_Kingdom_foot-and-mouth_outbreak
Foot and mouth disease had previously escaped a Kansas lab in 2004, not once but twice!
https://en.wikipedia.org/wiki/Plum_Island_Animal_Disease_Center#Laboratory_accidents
Ebola, Zika, H5N1, and Dengue have all escaped labs. People died.
https://t.co/bAXN0KjwNe?amp=1
Qué información proporciona la secuenciación del genoma del coronavirus SARS-CoV-2-The proximal origin of SARS-CoV-2
- https://notistecnicas.blogspot.com/2021/05/que-informacion-proporciona-la.html
- https://notistecnicas.blogspot.com/2021/02/que-es-la-secuenciacion-genetica-una.html
- https://notistecnicas.blogspot.com/2021/05/virus-que-se-usan-para-curar.html
- https://notistecnicas.blogspot.com/2020/12/los-virus-tambien-pueden-ser-los-buenos.html
- https://notistecnicas.blogspot.com/2021/02/la-oms-y-china-concluyen-que-el.html
- https://notistecnicas.blogspot.com/2020/03/un-estudio-en-2007-advirtio-de-que-los.html
- https://notistecnicas.blogspot.com/2020/07/el-papel-de-la-bioinformatica-en-el.html
- https://notistecnicas.blogspot.com/2021/03/la-necesidad-de-crear-una-agencia-one.html
10-Más sobre este tema:
Análisis estadístico de una secuencia de
ADN ()
Búsqueda de genes en una secuencia de ADN ()
Sequence Manipulation Suite (SMS): Random DNA Sequence - DNA stats -
Generación de secuencias aleatorias (Molbiotools): Random Sequence Generator
Análisis estadístico: Genomatix - Wordcount - Compseq
Enzimas de restricción: WebCutter - REBsites
Diseño de cebadores: Primer3 (Simgene) - Primer3
ORF: ORF Finder (NCBI) - ORF Finder (SMS)
Regiones codificantes: Codon Usage (SMS) - TESTCODE - tcode (EMBOSS) - FRAMEPLOT -
Regiones codificantes: Codig Potential Calculator - Coding Potential Assesment Tool
Bases de datos: Nucleotide
http://www.ehu.eus/biofisica/
https://www.ehu.eus/es/home
http://www.ehu.eus/biofisica/juanma/bioinf/covid_19.htm#covid_19
La secuenciación completa del virus permite analizarlo con profundidad, sus características, los pequeños cambios que determinan su comportamiento, su circulación y su difusión. De hecho, se han usado nuevos procesos de análisis bioinformático que han permitido llevar a cabo la secuenciación del genoma del coronavirus.
https://www.esciupfnews.com/2020/04/08/la-secuenciacion-del-coronavirus-sars-cov-2/
https://www.nationalgeographic.com.es/ciencia/como-pasan-coronavirus-animales-a-humanos_15392
https://www.ncbi.nlm.nih.gov/orffinder/
https://www.genomatix.de/cgi-bin/tools/tools.pl
https://www.bbc.com/mundo/noticias-51946236
https://www.elsevier.es/es-revista-revista-argentina-microbiologia-372-articulo-aplicacion-secuenciacion-masiva-bioinformatica-al-S0325754119300811
https://as.com/deporteyvida/2020/10/21/portada/1603269628_843264.html
https://coddii.org/tag/bioinformatica
No hay comentarios:
Publicar un comentario