Las mutaciones del coronavirus SARS-CoV-2
Por Francisco R. Villatoro, el 7 marzo, 2020. Categoría(s): Ciencia • Medicina • Noticias • Science • Virología ✎ 5
Quizás has leído por ahí que hay dos linajes (o cepas) del
coronavirus SARS-CoV-2, llamadas L y S, que solo se diferencian en un
único aminoácido mutado; S sería ancestro de L y ésta se transmitiría
más fácilmente (sería prevalente en el ~70% de los casos). En realidad,
lo único que hacen los virus de ARN es replicarse y mutar; por ello toda
persona infectada con el coronavirus SARS-CoV-2 tendrá en su interior
genomas víricos con diferentes mutaciones. Entre el 29 de noviembre de
2019 y el 7 de marzo de 2020 se han secuenciado 209 genomas completos de
SARS-CoV-2, en los que se han observado 111 mutaciones de aminoácidos
no sinónimas (recuerda que el código genético es redundante y las
mutaciones ocurren en los nucleótidos, luego hay muchas más mutaciones
de nucleótidos). Ninguna de estas mutaciones de aminoácidos tiene
relevancia clínica demostrada en la infección por COVID-19; luego no se
puede afirmar que haya dos cepas (o más) bien separadas. Así que no te
dejes engañar, por ahora solo existe una cuasiespecie de coronavirus
SARS-CoV-2 entre la población humana.
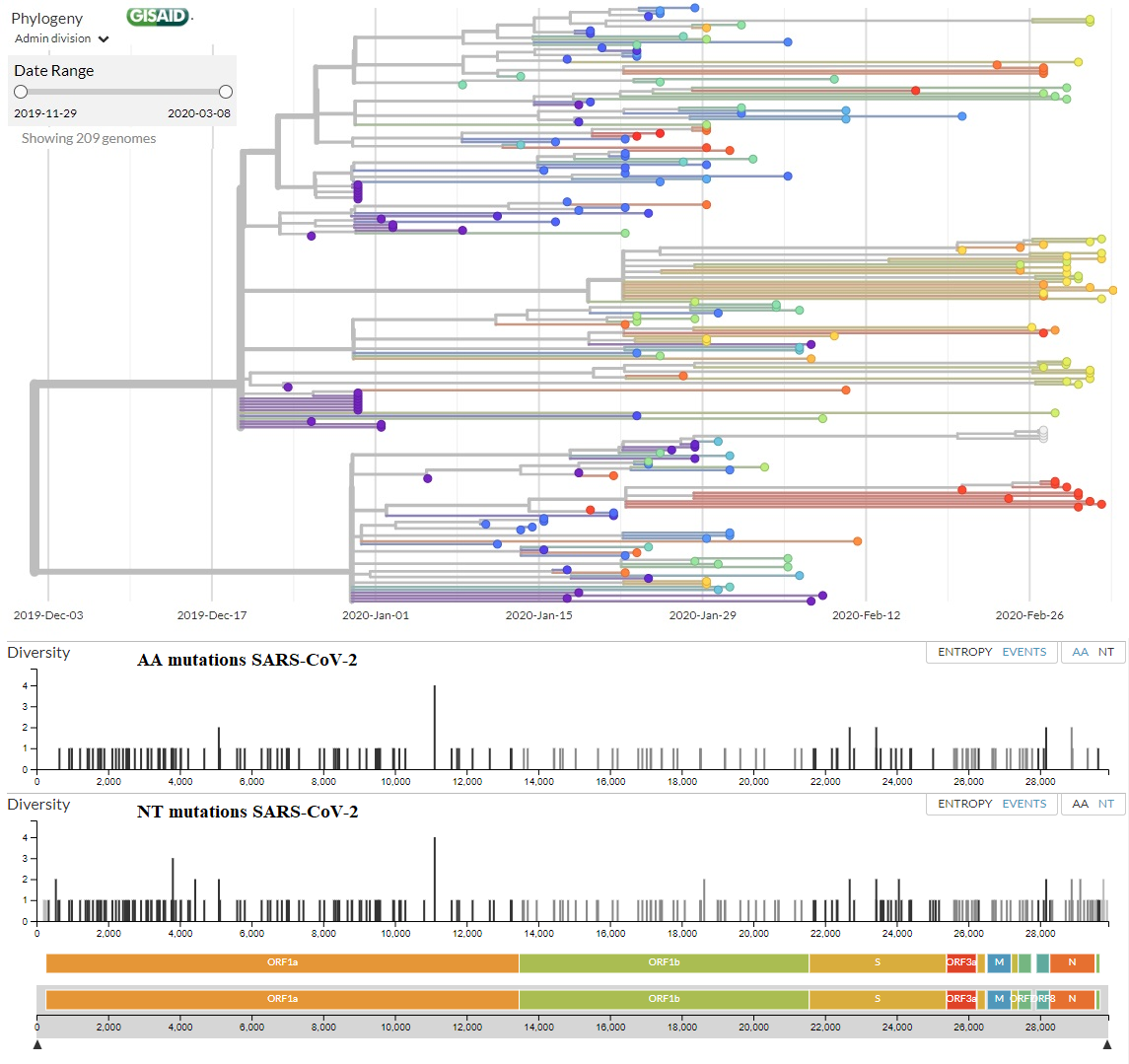
En esta figura te muestro el árbol filogenético (a fecha de 7 de
marzo de 2020) con los 209 genomas secuenciados (la última versión
actualizada la tienes en la web de NextStrain.Org, a partir de todos los genomas completos disponibles en la web de GISAID).
En la parte inferior tienes todas las mutaciones de aminoácidos (AA
mutations) y de nucleótidos (NT mutations) que se han encontrado, justo
encima de los genes del coronavirus donde se han encontrado (puedes leer
una explicación del genoma del SARS-CoV-2 en este blog en LCMF, 25 ene 2020).
En el eje vertical tienes la diversidad de nucleótidos (Diversity) que
mide el número promedio de diferencias entre nucleótidos en un sitio
determinado entre dos secuencias ADN para todos los pares posibles en la
población (más detalles y la fórmula que la calcula en la wikipedia). El listado de todas las mutaciones no sinónimas (de aminoácidos) lo tienes aquí, junto con el número de secuencias que la presentan.
Más información en Oscar A. MacLean, Richard Orton, …, David L.
Robertson, “Response to “On the origin and continuing evolution of
SARS-CoV-2”,” Virological.Org, 5 Mar 2020,
quienes critican el artículo con la propuesta de las dos cepas de
Xiaolu Tang, …, Jie Cui, Jian Lu, “On the origin and continuing
evolution of SARS-CoV-2,” National Science Review, nwaa036 (03 Mar
2020), doi: https://doi.org/10.1093/nsr/nwaa036.
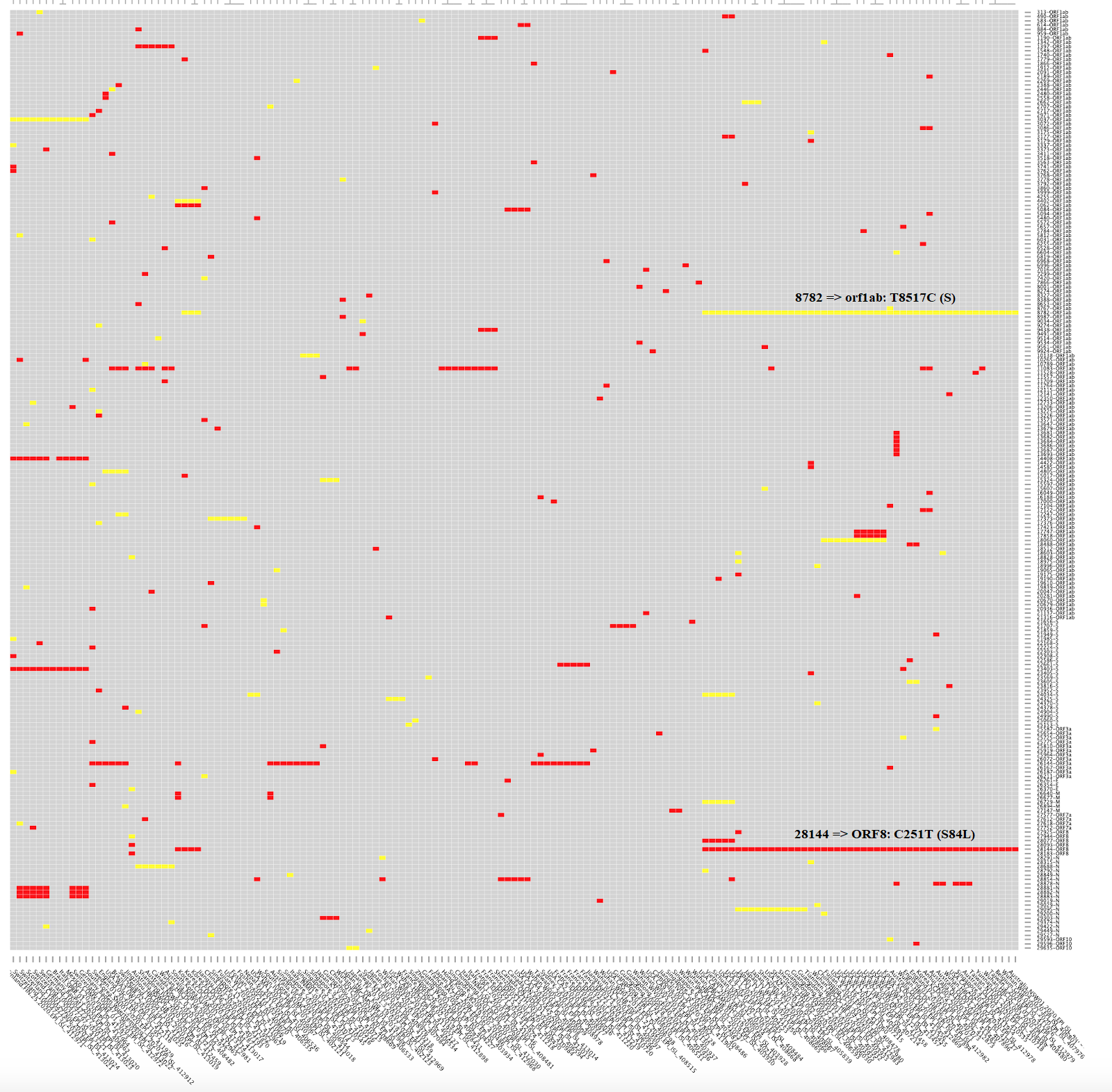
La crítica aún no publicada de MacLean et al. a la propuesta del
artículo publicado por Tang et al. parece obvia para quien es consciente
de que los coronavirus, como todos los virus de ARN, mutan mucho.
Máxime cuando la propuesta es que la cepa S sea ancestro de la cepa L y
que esta última se transmite más fácilmente (por ello, el ~70% de los
infectados la tendría). Sin embargo, ambas cepas solo se diferenciarían
en dos SNPs (se pronuncia esnips), polimorfismos de un solo
nucleótido. En concreto, un SNP sinónimo en la posición 8782 del ARN, en
el gen orf1ab, el cambio de una timina por una citosina en la posición
8517 de dicho gen (T8517C), que cambia el codón AGT de una serina por
AGC también de una serina; y otro SNP no sinónimo en la posición 28144
del ARN, en el gen ORF8, el cambio de una citosina por una timina en la
posición 251 de dicho gen (C251T), que cambia el codón de una serina por
el de una leucina en la posición 84 (S84L). Esta figura muestra las 111
mutaciones no sinónimas en rojo y las restantes mutaciones sinónimas en
amarillo; he aclarado dónde se encuentran las mutaciones 8782 (orf1ab:
T8517C) y 28144 (ORF8: C251T, S84L).
La teoría evolutiva predice que el coronavirus SARS-CoV-2 está
sometido a mutaciones continuas que producen una deriva genética que
acabará conduciendo a su separación en diferentes cepas en el futuro. Lo
habitual es que las cepas que mejor se adapten al ser humano (es decir,
las que tengan una letalidad reducida y produzcan síntomas más leves)
sean las que se acaben propagando con mayor facilidad. Pues al
coronavirus le «interesa» sobrevivir el máximo tiempo posible entre los
humanos y que no le pase lo que pasó con el SARS-CoV (cuya epidemia fue
contenido y desapareció de la circulación entre los humanos).
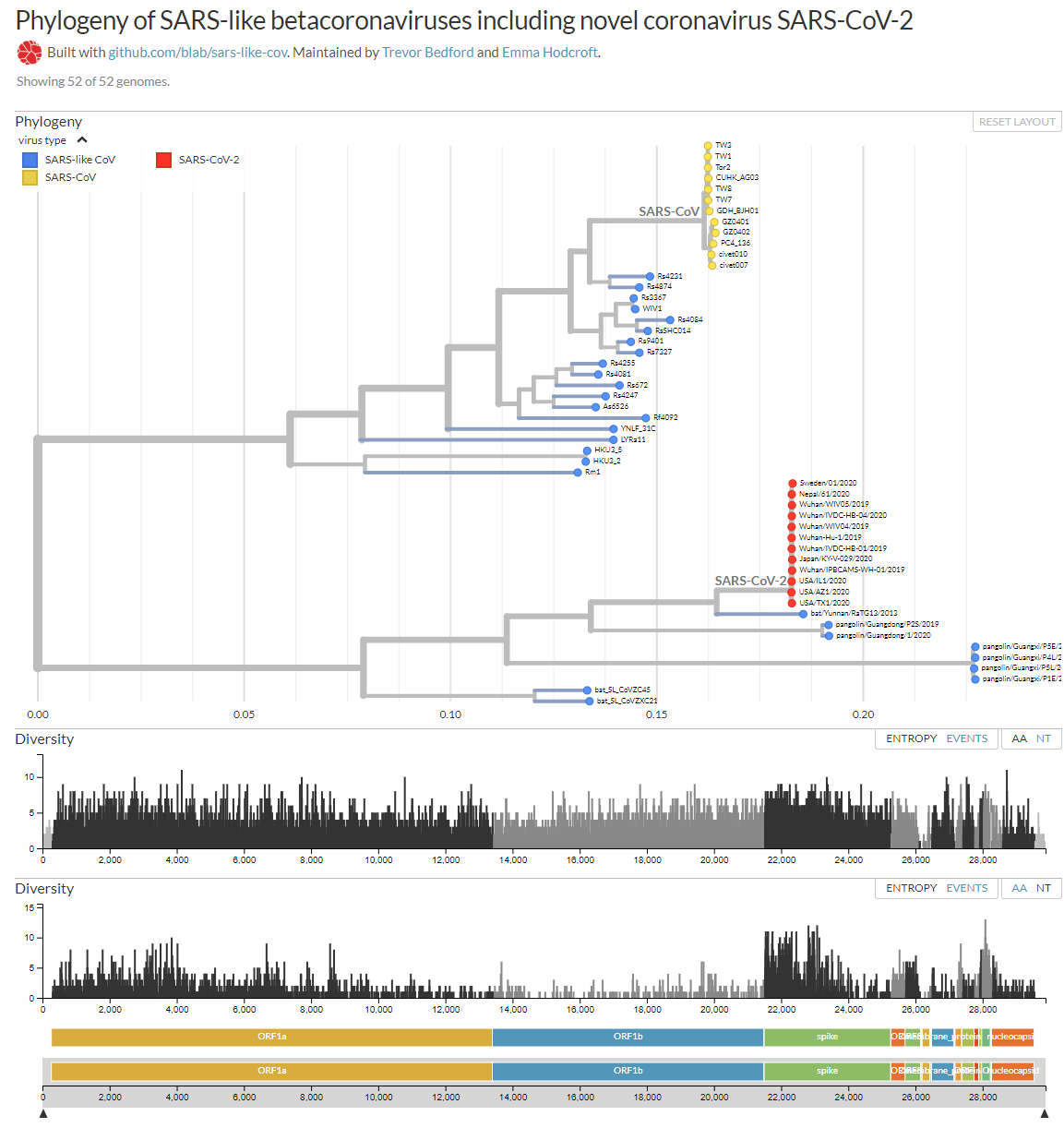
Y, por cierto, seguro que en Twitter y en medios sensacionalistas has
oído que el coronavirus SARS-CoV-2 podría haber escapado del
laboratorio BSL-4 de Wuhan. Nada más lejos de la realidad. Se han
publicado varios artículos que desmontan esta conspiración y demuestran
que la variación genética natural de los betacoronavirus es suficiente
para explicar la aparición de nuevos virus que puedan infectar a humanos
(como ocurrió con SARS y MERS, y ahora con COVID-19). Lo que pasa es
que las noticias virales como las del coronavirus acaban generando de
manera espontánea gran número de conspiraciones, porque todos tenemos un
poco de conspiranoicos aunque nos pese. Te recomiendo leer a Daniel
Jolley, Pia Lamberty, El coronavirus es un campo abonado para los
‘conspiranoicos’,” The Conversation, 05 mar 2020.
Me permito aportar mi granito de arena con esta figura que te muestra
el arból filogenético de los betacoronavirus similares a SARS, entre
ellos SARS-CoV-2, así como las mutaciones que se han observado en sus
ARN. Prácticamente todos los nucleótidos (NT) han mutado alguna vez; y
casi todos los aminoácidos (AA) también. Como se dispone de más genomas
de los que infectan a humanos (SARS-CoV en amarillo y SARS-CoV-2 en
rojo) que de los demás, se observa que la diversidad genética es mucho
mayor en la glicoproteína S, responsable de la infección a humanos; pero
su razón de ser es un simple sesgo estadístico. Como nos recuerda
Ignacio López-Goñi en Twitter:
“No, no se ha escapado de un laboratorio, ni es un arma biológica, la
naturaleza se basta y se sobra para generar nuevos virus”.
En resumen, el coronavirus SARS-CoV-2 está mutando de forma continua
como cuasiespecie que es, pero todavía no hay datos suficientes para
afirmar que haya más de una cepa. Que no te engañen ni te asusten con
supuestas mutaciones que incrementan su letalidad; lo más habitual es
que las mutaciones más letales, si algún día llegan a aparecer, no se
propaguen entre la población. Lo peor que nos puede pasar es que el
virus se acomode a los humanos y se haga estacional como el virus de la
gripe; en dicho caso tendremos epidemias de COVID-19 todos los años. Por
fortuna, habrá vacunas y en poco tiempo todo el mundo se olvidará de su
existencia (como han hecho con el virus de la gripe A). Eso sí, mucha
gente en grupos de riesgo no se vacunará (como no se vacunan de la
gripe) y fallecerá (como fallecen por la gripe), pero sus decesos ya no
serán noticia en los medios.
- https://francis.naukas.com/2020/03/07/las-mutaciones-del-coronavirus-sars-cov-2/?fbclid=IwAR3s8JogiYdsoQOxKmiD5zFhaNpzltGvB0Qhp6VwDNg_ovQwPw3TnT_iKAk
La teoría
evolutiva predice que el coronavirus SARS-CoV-2 está sometido a
mutaciones continuas que producen una deriva genética que acabará
conduciendo a su separación en diferentes cepas en el futuro. Lo
habitual es que las cepas que mejor se adapten al
ser humano (es decir, las que tengan una letalidad reducida y produzcan
síntomas más leves) sean las que se acaben propagando con mayor
facilidad. Pues al coronavirus le «interesa» sobrevivir el máximo tiempo
posible entre los humanos y que no le pase lo que pasó con el SARS-CoV
(cuya epidemia fue contenido y desapareció de la circulación entre los
humanos).
-https://cmmid.github.io/topics/covid19/severity/diamond_cruise_cfr_estimates.html?fbclid=IwAR0sddz4uhx6YHhc_vjr_WtiCJuS2bWXzlWq2M42MNShGun5KS41p-QE-os
No hay comentarios:
Publicar un comentario