¿Por qué necesitábamos un pangenoma humano?
Lluis Montoliu, el 21 mayo, 2023. Categoría(s): diagnóstico genético • genética • genoma • pacientes ✎
Son ya diversas las entradas que he dedicado al genoma humano en este blog sobre Gen-Ética. En particular dediqué una entrada a explicar cómo utilizábamos nuestro genoma de referencia y la influencia del mismo a la hora de encontrar mutaciones causantes de enfermedad, por ejemplo. Frecuentemente también me he referido al genoma humano como un puzle, un rompecabezas de muchas fichas, tantas como letras (o bases) tiene nuestro genoma. Esa metáfora me sirvió para explicar la adición de unas 200 millones de letras adicionales que se añadieron recientemente, completando la imagen del puzle inicial de referencia, al cual le faltaba rellenar todavía algunos agujeros. Pues ahora tenemos que complicar un poco más la visión e idea que tenemos de nuestro genoma. Seguid leyendo y descubriréis por qué necesitábamos el pangenoma y por qué tendremos que seguir trabajando para que progresivamente sea capaz de capturar la diversidad genética que tenemos los seres humanos.
Todos los seres humanos nos parecemos, aproximadamente, al 99,9%. Es decir, cada par de seres humanos nos diferenciamos solamente en un 0,1%. Esto puede parecer un número pequeño pero en realidad son millones de letras las que tenemos diferentes las personas, comparadas dos a dos. Si consideramos 3.000 millones de pares de letras el tamaño del genoma humano (que en realidad serían 3.200 millones de pares de letras con la reciente actualización, y si tenemos en cuenta que cada uno de nosotros heredamos la mitad de nuestro genoma de nuestro padre y la otra mitad de nuestra madre, es decir, 3.200 millones de pares de letras de nuestra madres y otros tantos de nuestro padre, 6.400 millones pares de letras en total) entonces el 0.1% de esta cifra enorme es un número nada desdeñable: 6,4 millones de pares de letras. En realidad son entre 3 y 6 millones de letras las que hay de diferencia entre cada ser humano. Y, obviamente, en función del genoma que usemos como referencia podremos asignar una determinada letra en una determinada posición como idéntica o diferente a la del genoma de referencia. Si cambiamos la referencia puede cambiar la consideración de esta letra, y pasar de ser una mutación (al ser distinta de la letra en el genoma de referencia) a ser idéntica. No puede haber un solo genoma que actúe de referencia para todos los seres humanos.
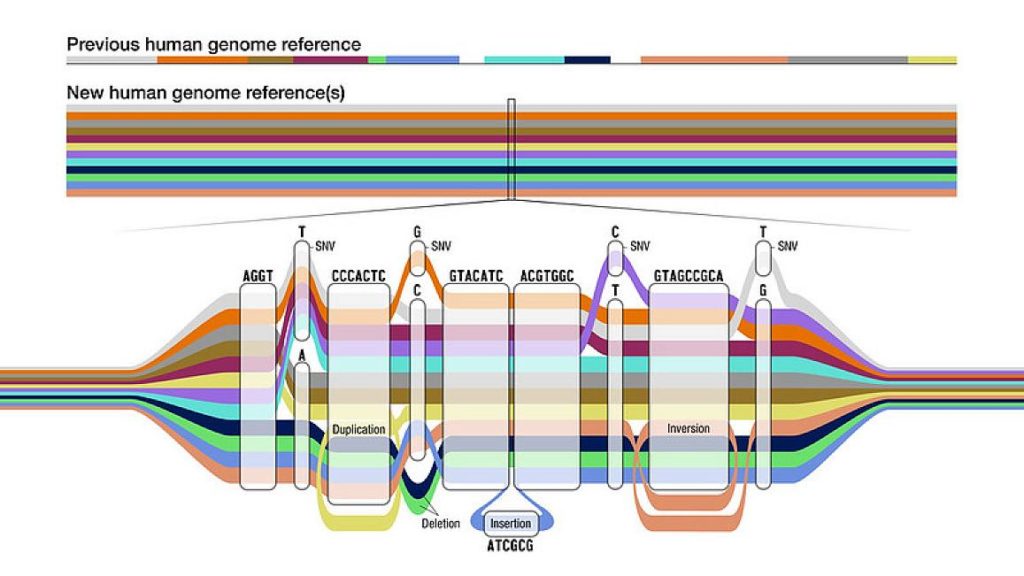
Pero es que además nuestros genomas no solo difieren en posiciones individuales de las letras. También se diferencian en fragmentos cromosómicos que están a veces duplicados, una o múltiples veces, o invertidos. O las dos cosas. O en inserciones de fragmentos de ADN de tamaño variable. O por lo contrario, en deleciones, pequeños o grandes segmentos cromosómicos que faltan en alguna persona, mientras que sí están presentes en otras. Todo esto complica sobremanera el diagnóstico genético de los pacientes, la búsqueda de la causa molecular. Y aquí sí que podemos entender fácilmente que será todavía más complejo establecer dónde está y qué es una mutación, y si determinado cambio de letra es un mutación o es una variante común de este grupo de personas que viven en un mismo entorno geográfico. Según el genoma de referencia que escojamos concluiremos que aquella letra es habitual o, por el contrario, es distinta y por ello la debemos considerar una mutación. Observad atentamente el siguiente gráfico, que creo ayudará a entender esta cuestión.

Somos casi 8.000 millones de personas en este planeta. Y cada uno de nosotros tenemos un genoma ligeramente distinto. Esencialmente similar, en lo fundamental (de ahí que compartamos el 99,9%) pero con suficiente espacio para la diversidad genética en esos pocos millones de letras que nos diferencian y distinguen cuando comparamos nuestro genoma con el de cualquier otro ser humano. Y da igual el aspecto externo que tengamos y el color de nuestra piel, ojos o cabellos, como ya expliqué en mi libro Genes de colores. Existen variantes genéticas, mutaciones, que aparecen en una población, en un grupo de personas de una determinada ubicación geográfica, y se expanden hasta hacerse comunes en ese grupo de personas. Otros grupos habrán incorporado otras variantes. Por ello, cuando intentamos buscar la causa genética de una enfermedad en un paciente y necesitamos comparar su genoma con uno de referencia lo que deberíamos hacer es usar el genoma de referencia más parecido al del grupo de personas al que pertenece el paciente. Si usamos otro genoma de referencia es posible que detectemos otras mutaciones que, en ese grupo inicial no son tales, mientras que pueda pasar desapercibida la variante que en definitiva es la causante de la enfermedad congénita.
Idealmente nos encantaría poder tener el genoma de todos y cada uno de los habitantes de la Tierra, con objeto de obtener y archivar toda la complejidad y diversidad genética humano. Esto es actualmente imposible, inviable, irrealizable. ¿Pero cuál sería una aproximación válida para intentar acercarse a este objetivo útopico? Pues intentar obtener el genoma de diferentes grupos humanos, tantos como fuéramos capaces de identificar y secuenciar, para poder usar esta información para comparar el genoma de un paciente de un determinado grupo humano con el genoma de referencia que le correspondiera, y poder asegurar, con mayor fiabilidad, qué es una mutación y qué no lo es en ese conjunto de seres humanos. Pues esto es, ni más ni menos, lo que han hecho un conjunto de investigadores del consorcio internacional «Human Pangenome Reference Consortium» obteniendo los genomas de 47 personas representativas de otros tantos grupos humanos y con el objetivo de aumentar este número hasta 350 en 2024. Los resultados los han publicado en un conjunto de artículos publicados en la revista científica Nature.
El genoma que usamos actualmente como referencia (que se obtuvo a partir de secuencias de cromosomas obtenidas de diferentes células de diferentes individuos) no existe como tal ni ha existido nunca en un ser humano. Es un collage, un compendio de secuencias, un mosaico de letras obtenidas de hasta 20 personas distintas, de las cuales una de ellas ha contribuido con hasta el 70% del total del genoma. En otras palabras, el genoma de referencia que hemos usado hasta ahora es una mezcla de secuencias arbitrarias que nunca coexistieron en una persona. Por lo tanto no debería sorprendernos las dificultades que tenemos de diagnosticar muchas personas (hasta un 30% de personas con alguna enfermedad rara congénita, en las que somos incapaces de encontrar la o las mutaciones causales) dado que es probable que la secuencia que contenga la mutación en el paciente no estuviera representada en ese genoma mosaico de referencia o, por el contrario, estuviera anotada como secuencia común, correcta, y no pudiéramos detectarla como mutación en el paciente. En definitiva, un despropósito. Un enorme problema que requería una solución. De ahí la gran aportación que significa disponer no de un genoma de referencia sino de un PANGENOMA, que actualmente recoge la diversidad genética de 47 personas, representativas de 47 poblaciones humanas diferentes.
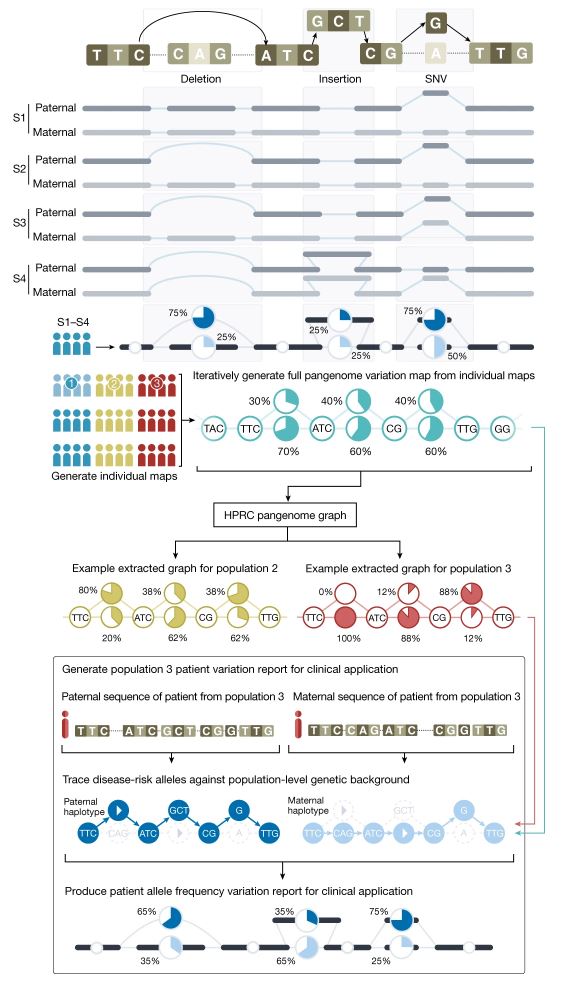
Para transformar toda esta ingente fuente de variabilidad genética en algo gestionable este consorcio del pangenoma tuvo que desarrollar diferentes estrategias para asegurarse el poder obtener secuencias de calidad monoparentales (o bien la secuencia de la madre o la del padre, gestionadas por separado) y para graficarlas de forma inteligible e interpretable. La figura adjunta, del artículo principal del consorcio, ilustra la aproximación experimental que tomaron estos autores y su resultado. Para empezar no usaron las técnicas de secuenciación genómica masiva habituales, que producen millones de lecturas de secuencias de pequeño tamaño, habitualmente entre 50 y 300 letras de longitud, que pueden ser muy difíciles de cartografiar y ensamblar, si están incluidas dentro de segmentos repetitivos. Por el contrario usaron tecnologías (como PACBIO) que permiten obtener segmentos de más de 10.000 letras contiguas con alta fiabilidad y que pueden asignarse a uno de los dos progenitores (recordemos que todo ser humano portamos en realidad dos genomas, la mitad del genoma heredado de nuestro padre y la otra mitad heredado de nuestra madre). Para establecer lo que era secuencia paterna y materna procedieron, para cada individuo, a secuenciar TRÍOS, es decir: padre, madre e hijo/a. Esta aproximación permite establecer, de forma inequívoca, si una secuencia es de origen paterno o materno. La representación gráfica del pangenoma nos permite reconocer, en porcentajes, que distintas secuencias coexisten para una misma ubicación del genoma humano.

Recuperando el ejemplo de los puzles, el pangenoma vendría a representar que en un lugar de un puzle único con una sola foto de referencia tuviéramos 47 puzles distintos, con 47 fotografías de referencia ligeramente distintas, cada uno con un número ligeramente distinto de piezas (en unas se añadirían piezas, en otros faltarían piezas, en otros se cambiaría alguna pieza por otra, en otros se duplicarían algunas de las piezas….). Y así, a la hora de construir un puzle y tomar una de las fotos de referencia para localizar la posición de cada una de las piezas sería muy importante decidir qué fotografía de qué caja tomamos como referencia, para que no nos faltaran/sobraran piezas y para que (asumiendo que una persona enferma sería una persona con una pieza incorrecta, o de más o de menos) fuéramos capaces de localizar la pieza incorrecta que sería la que explicaría el problema de ese puzle, la causa genética de la enfermedad de ese paciente.
Y así fue como este consorcio ha obtenido este PANGENOMA, por el momento basado en 47 personas diferentes pero que en un futuro próximo aumentará su complejidad hasta llegar a 350 personas distintas. En palabras de los autores el pangenoma es «the collective whole-genome sequences of multiple individuals representing the genetic diversity of the species», las secuencias colectivas del genoma completo de múltiples individuos que representan la diversidad genética de la especie. Toda la información actualmente disponible sobre el PANGENOMA puede obtenerse a partir de las publicaciones referidas en la revista Nature o a través de su portal web.
Son igualmente interesantes los comentarios sobre el PANGENOMA de Marc Martí-Renom (CRG, CNAG) y Gemma Marfany (UB, CIBERER) en SMC ES, el artículo de Antonio Regalado en el MIT Technology Review, y la entrevista que nos hizo Lorenzo Milá a Belén Pérez (CBMSO-UAM/CSIC, CIBERER, CEDEM) y a mi para su programa Objetivo Planeta de RTVE.
https://montoliu.naukas.com/2023/05/21/por-que-necesitabamos-un-pangenoma-humano/










/cloudfront-eu-central-1.images.arcpublishing.com/prisa/ARLYK4WNM5CYNPIUMQEFBGODDI.jpg)





