La vida se abre camino: ¿cómo evoluciona una bacteria diseñada en el laboratorio con un genoma mínimo? Se puede simplificar el genoma hasta lo esencial, pero eso no detiene a la evolución
La vida se abre camino
Estudian cómo evoluciona una bacteria diseñada en el laboratorio con un genoma mínimo
Quizá recuerdes esta frase mítica: “Si algo nos ha enseñado la historia de la evolución es que la vida no puede contenerse. La vida se libera, se extiende a través de nuevos territorios y rompe las barreras dolorosamente, incluso peligrosamente, … la vida se abre camino”. La dijo Ian Malcolm, el personaje de Jeff Goldblum, en la película de ciencia ficción Jurassic Park de 1993.
En una célula, a lo largo del tiempo, se van acumulando cambios o mutaciones en el genoma, que, si suponen alguna ventaja, son seleccionadas y pasan a la descendencia. Con el tiempo, las células pueden adquirir nuevas funciones y/o adaptarse a nuevos ambientes. La variabilidad genética y la selección natural son la base de la evolución celular. Para que la evolución “funcione” es necesario que la célula posea partes del genoma “redundantes” o extra que puedan acumular esas mutaciones, sin afectar a su viabilidad. Los genes esenciales para la supervivencia de la célula se mantienen (una mutación en un gen esencial podría llegar a ser letal para la célula), mientras que otras partes del genoma que no sean esenciales pueden actuar como fuentes de variabilidad y evolución. Así es cómo pensábamos que funcionaba la evolución y la selección natural… hasta ahora.
La vida, ¿siempre se abre camino? ¿qué es antes la vida o la evolución?
Ahora los investigadores han estudiado cómo se enfrenta a las fuerzas de la evolución una célula mínima modificada con el genoma más pequeño que existe, en comparación con la célula original de la cual deriva. Veamos cómo lo han hecho.
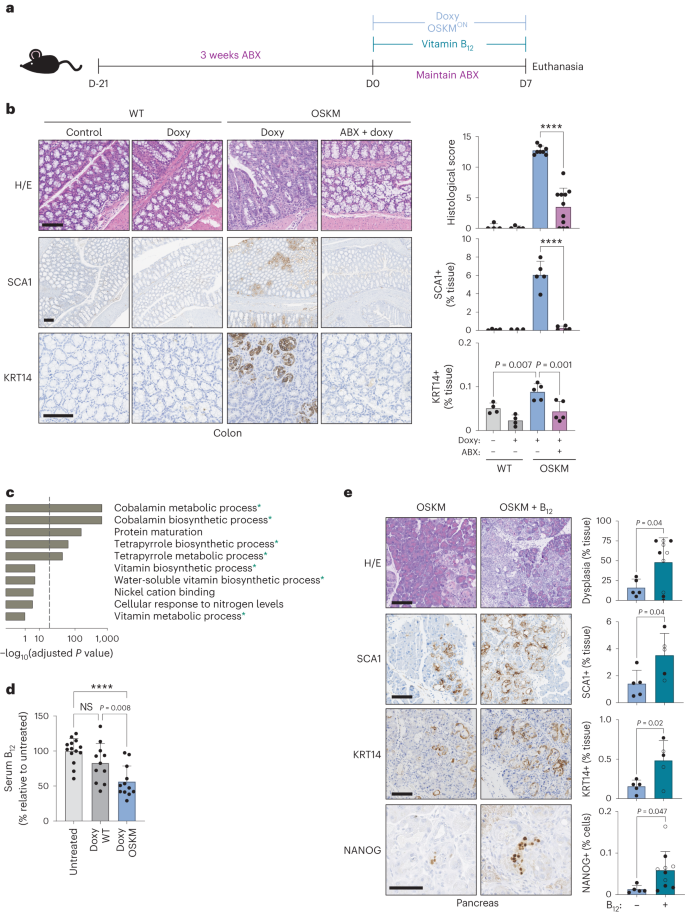
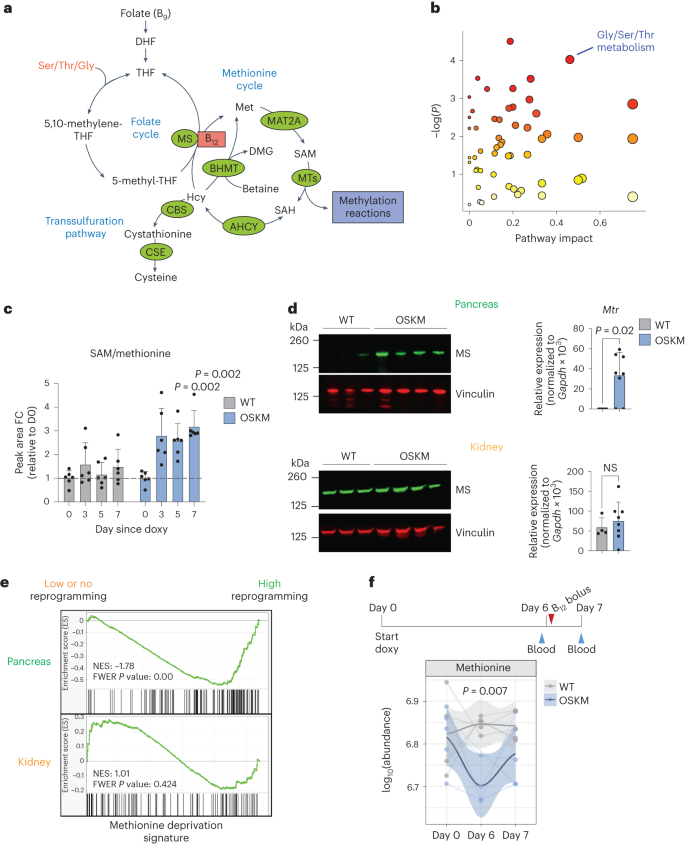
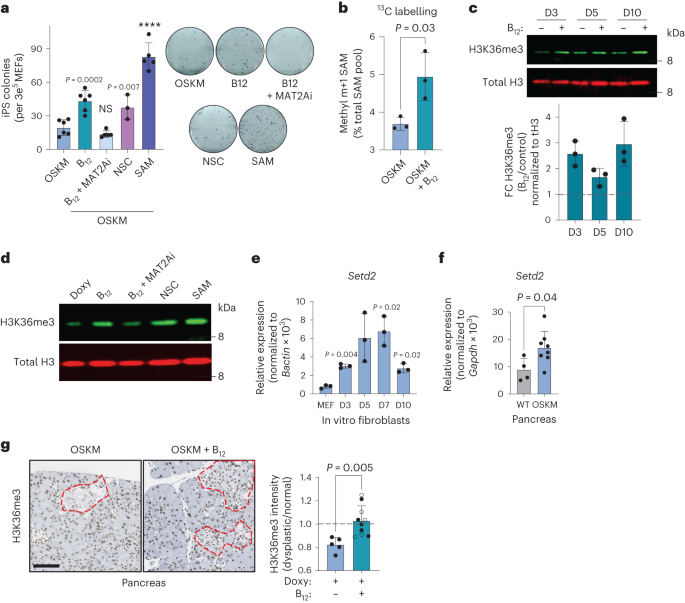
Hace ya varios años, investigadores del J. Craig Venter Institute en California diseñaron y sintetizaron el primer genoma bacteriano mínimo. Para ello, emplearon la bacteria Mycoplasma mycoides, un patógeno del ganado y pequeños rumiantes que puede causarles una enfermedad respiratoria. El género Mycoplasma son bacterias muy pequeñas (0,2-0,3 micras) que carece de pared celular y con un genoma también muy pequeño. En este trabajo eliminaron el 47% de los 901 genes del genoma natural de Mycoplasma mycoides, reduciendo el genoma al conjunto de genes más pequeño necesarios para la vida autónoma de una célula. Con 473 genes, el genoma mínimo artificial de esta bacteria, que pasó a denominarse M. mycoides JCVI-syn3B, es el genoma más pequeño de un organismo autónomo de vida libre. Es decir, con un solo gen menos, esa bacteria ya no puede desarrollarse. M. mycoides JCVI-syn3B es, por tanto, una versión sintética minimizada de la bacteria M. mycoides. Curiosamente, 149 de esos 473 genes tenían una función biológica desconocida. (Para que te hagas una idea, otras bacterias como Bacillus subtilis o Escherichia coli tiene entre 4.000 y 5.000 genes).
Aunque M. mycoides JCVI-syn3B podía crecer y dividirse en condiciones de laboratorio, los investigadores querían saber cómo respondería a la evolución a lo largo del tiempo esta célula artificial con un genoma mínimo. Como hemos comentado, cada gen en su genoma es esencial, por lo que la mayoría de los cambios o mutaciones afectarían a funciones esenciales y, en principio, serian letales. Se podría pensar que en esta célula mínima no hay margen para mutaciones, lo que limitaría su capacidad de evolucionar. Este microorganismo mínimo debería tener en teoría una muy baja capacidad de adaptación. Pero ¿qué sucede si dejas que este organismo se reproduzca por sí mismo a lo largo de muchas generaciones? Con el tiempo, ¿se seleccionarían algunas mutaciones?, ¿mejoraría su capacidad de adaptación?
Dicho y hecho. Los investigadores han dejado evolucionar a M. mycoides JCVI-syn3B durante 2.000 generaciones para ver qué sucedía (algunos han hecho el cálculo y dos mil generaciones bacterianas equivalen aproximadamente 40.000 años de evolución humana). Y lo que sucede es «mucho». Algo que parece ser una constante evolutiva es que, a menor tamaño del genoma, mayor es la tasa de mutación, y M. mycoides JCVI-syn3B mostró la tasa más alta de mutación jamás medida. Esto en realidad tiene sentido, porque en el proceso que llevó a su síntesis se eliminaron los genes necesarios para corregir los errores en la replicación del ADN y reparar mutaciones.
Además, el 80% de las mutaciones en M. mycoides JCVI-syn3B fueron puntuales, cambios de un solo nucleótido. Sin embargo, a diferencia de su progenitor, las células mínimas de M. mycoides JCVI-syn3B mostraron una preferencia por los cambios de Guanina a Citosina y de Adenina a Timina (por el contario en el M. mycoides original las mutaciones son en la otra dirección, de Citosina a Guanina y de Timina a Adenina).
Al comparar las células al cabo de 2.000 generaciones con las del inicio, encontraron que las células mínimas M. mycoides JCVI-syn3B se adaptaron aproximadamente un 40% más rápido, pero terminaron al mismo nivel que las M. mycoides originales. Parece ser que simplificar el genoma no debilitó a la célula como para que no pudiera adaptarse. Lo que sí cambiaron fueron varios genes necesarios para la biosíntesis de los lípidos, lo que puede estar relacionado con cambios en la membrana celular.
Restringir el genoma puede tener consecuencias en el tamaño celular
Otro cambio significativo tiene que ver con el tamaño celular. Mientras que las células originales de M. mycoides aumentaron su diámetro en un 85% y su volumen en 10 veces a lo largo de las 2.000 generaciones, las células mínimas de M. mycoides JCVI-syn3B no cambiaron de tamaño durante todo el experimento. Quizá esto tiene que ver con el control celular de la relación superficie/volumen: al aumentar de tamaño tienes más espacio para almacenar proteínas, lípidos y nutrientes, pero empeora la relación superficie/volumen y se dificulta el transporte de nutrientes dentro y fuera de la célula. Las células mínimas quizá sean incapaces de transportar suficientes nutrientes para construir una célula más grande.
Se podría pensar que la reducción del genoma en una célula mínima podría llevar a la extinción a lo largo de las generaciones. Pero no es eso lo que ha ocurrido. El que las funciones celulares básicas se mantengan a lo largo del tiempo es importante cuando se utilizan este tipo de células mínimas en biotecnología. La selección natural durante el crecimiento prolongado en el laboratorio (2.000 generaciones) compensó cualquier efecto perjudicial de la reducción del genoma. Por tanto, se puede simplificar el genoma celular hasta lo esencial, pero eso no detiene a la evolución. La célula mínima sintética puede evolucionar tan rápido como una célula normal. Esto demuestra la tremenda capacidad que tiene los seres vivos para adaptarse, incluso cuando poseen un genoma artificial con el mínimo número de genes necesarios para sobrevivir.
Este artículo me ha recordado una conversación con un buen amigo que un día me preguntó ¿Qué es antes la vida o la evolución? Sin dudarlo contesté: La evolución, sin evolución no hay vida. Después de leer este artículo… tengo dudas.
Evolution of a minimal cell. R. Z. Moger-Reischer, y col. Nature. 2023. 620:122–127.
Design and synthesis of a minimal bacterial genome. Clyde A. Hutchison III, y col. SCIENCE. 2016. 351(6280).
https://microbioblog.es/la-vida-se-abre-camino