Desde 2006 ya se sabía que los coronavirus ingresan al cerebro e inducen diferentes tipos de daño inflamatorio e inmunológico, entonces como es posible que 14 años después dejen que el #SARSCoV2 corra libremente entre la población sin ningún tipo de control

Coronavirus infection of the central nervous system: host–virus stand-off
Nature Reviews Microbiology volume 4, pages 121–132 (2006)
The paper below was published in 2006, way before #SARSCoV2. It is a detailed analysis of how different coronaviruses enter the brain and induce different types of inflammatory and immunological damage. We know exactly what happens in the brain during the acute and chronic phases of coronavirus infection: - Viral antigen (vAg) and viral RNA (vRNA) levels decrease in the brain after two weeks (that's why it is so hard to detect active viral presence in human postmortem tissue). - We know that following the reduction of the viral presence in the brain comes primary demyelination, which is most severe between 2 and 3 weeks after infection. - We also know precisely the kinetics of the most common pro-inflammatory cytokines involved in the acute response to the viral presence in the brain. Their early detection can help prevent late damage and #LongCovid. Yet, after years of #COVID19, we are still debating if SARS-CoV-2 enters the brain (all the other known human coronaviruses do), and we still don't have early markers and treatments to prevent long-term neurological damage after infection. How is this possible?
https://twitter.com/DaniBeckman/status/1741170872210100697
Coronavirus infection of the central nervous system: host–virus stand-off
Key Points
-
Coronaviruses infect humans, rodents and several agriculturally important animals.
-
Mouse hepatitis virus (MHV) causes acute infections of the murine liver and lungs and persistent infections of the gastrointestinal tract and central nervous system (CNS).
-
Interactions of immune effectors and cells of the CNS can be studied using a non-lethal gliatropic strain of MHV. This model sheds light on the interplay of cytokines, chemokines and innate and adaptive immune effectors during acute infection, as well as their role in regulating coronavirus persistence.
-
This review summarizes data that show how distinct phases of CNS infection are associated with the induction of innate danger signals, altered patterns of inflammatory cells and expression of antiviral effector functions. This leads to a state of virus–host coexistence that is beneficial to the survival of both.
-
During acute infection, the principal antiviral effectors are virus-specific T cells, which use distinct mechanisms to control virus replication in a CNS-cell-type specific manner. Control of viral replication in CNS-resident macrophages (microglia) and astrocytes is dependent on CD8+ T-cell perforin-mediated cytolysis. By contrast, control of replication in oligodendrocytes requires secretion of the soluble mediator, interferon-γ.
-
Control of virus replication is accompanied by downregulation of CD8+ T-cell cytolytic function and recruitment of virus-specific antibody-secreting cells into the CNS. Maintenance of local secretion of neutralizing antibody is crucial in preventing the re-emergence of infectious virus, indicating that virus persists in a replication-competent form.
-
In the MHV model, the limitation of prolonged cytolytic activity sustains CNS function, while soluble mediators control, but cannot eradicate, persistent infection.
Abstract
Varios virus infectan el sistema nervioso central (SNC) de los mamíferos, algunos con consecuencias devastadoras, mientras que otros provocan infecciones crónicas o persistentes infecciones crónicas o persistentes con poca o ninguna patología manifiesta. La infección del SNC murino por el coronavirus
del SNC murino ilustra la contribución tanto de la respuesta inmunitaria innata como de los efectores específicos del huésped. respuesta inmunitaria innata como de mecanismos efectores específicos del huésped que que controlan la replicación del virus en distintos tipos de células del SNC. A pesar de mediada por células T de la infección vírica aguda, los mecanismos reguladores del huésped del huésped, probablemente diseñados para proteger la integridad del SNC, contribuyen al fracaso en la eliminación del virus. A diferencia de los mecanismos efectores citolíticos citolíticos expresados durante la infección aguda, la inmunidad humoral no lítica prevalece en la supresión del virus infeccioso durante la persistencia.
Several viruses infect the mammalian central nervous system (CNS), some with devastating consequences, others resulting in chronic or persistent infections associated with little or no overt pathology. Coronavirus infection of the murine CNS illustrates the contributions of both the innate immune response and specific host effector mechanisms that control virus replication in distinct CNS cell types. Despite T-cell-mediated control of acute virus infection, host regulatory mechanisms, probably designed to protect CNS integrity, contribute to the failure to eliminate virus. Distinct from cytolytic effector mechanisms expressed during acute infection, non-lytic humoral immunity prevails in suppressing infectious virus during persistence.
Similar content being viewed by others

Main
Interactions between the immune system and the central nervous system (CNS) constitute the most complex and interactive regulatory network in mammals. The high degree of specialization of cell types that comprise the CNS, and their intricate communication, controls both cognitive and vital functions. Disruption of the communication network and poor CNS regenerative properties make this organ vulnerable to microbial as well as physical injury. Although it is known that host responses must be strictly regulated to preserve CNS function and to minimize the incidence of autoimmunity, the factors regulating CNS immune and repair responses are not well understood. In addition to the absence of a dedicated lymphatic drainage system, CNS cells express few, if any, molecules encoded by the major histocompatibility complex (MHC)1,2,3. Therefore, in the quiescent CNS there is little endogenous antigen presentation or potential to activate T cells. Although the underlying basis for this limited immunological activity is not completely understood, interactions between both neurons and the glial population represented by microglia, astrocytes and oligodendrocytes (Box 1), as well as constitutive secretion of neurotrophins and transforming growth factor-β, might contribute to this quiescent resting state1,4,5,6,7. The limited expression of adhesion molecules by endothelial cells of the blood–brain barrier (BBB) and the presence of tight junctions between these cells also limit or prevent large molecules, such as antibodies, and T cells from entering the CNS2,8. Despite this, a few activated/memory CD4+ and CD8+ T cells randomly patrol the CNS in the absence of 'danger' signals, and either exit or die in situ in the absence of antigen recognition2,8.
Mammals have evolved many immune effector mechanisms to eliminate pathogens that infect the CNS7,9. The vigorous inflammatory responses that are induced during many CNS infections contrast dramatically with its quiescent steady state. These inflammatory responses include rapidly induced, non-specific cellular and soluble effectors that provide an innate antimicrobial defence and facilitate development of antigen-specific effectors, which exert antimicrobial function and establish long-lived immunological memory. Some effectors mediate specific functions, whereas others mediate pleiotropic effects. Furthermore, several regulatory mechanisms limit immune responsiveness to avoid damage of uninfected host cells or the induction of autoimmunity10,11.
The conflicting needs for pathogen elimination and protection from cellular damage make the mammalian CNS a partially protected environmental niche that is a prime target for persistent viral infections. Viruses that persist in the human CNS include DNA viruses, as exemplified by herpes simplex virus and JC polyomavirus; RNA viruses, such as measles virus; and retroviruses, such as HIV and HTLV-1 (Refs 9,12–14). Several viruses that establish chronic infections in the rodent CNS provide useful models to examine both the roles and regulation of immune effectors in this vital organ. Collectively, these models have provided a wealth of information about the genetics of host resistance, acute and chronic viral infection as well as host defence mechanisms. Chronic, viral rodent CNS pathogens that are associated with myelin loss include two well characterized RNA virus models: Theiler's murine encephalomyelitis virus (TMEV), a member of the non-enveloped Picornaviridae, and mouse hepatitis virus (MHV), a member of the enveloped Coronaviridae. Although CD8+ T cells are important in controlling the acute phase of both infections, these viruses can escape immune surveillance and establish chronic CNS infection with ongoing myelin loss14,15,16,17,18.
Despite similar disease pathologies during chronic disease, infectious TMEV is present in the CNS during chronic disease19. By contrast, infectious MHV remains undetectable during persistence although MHV viral antigens and RNA are retained16,17,18. Another distinguishing characteristic during chronic TMEV infection is that chronic inflammation involves activation of self-reactive T cells20,21. By contrast, control of infectious MHV results in a slowly resolving, but chronic, CNS disease that is associated with minimal inflammation16,17,18. These chronic pathological changes in the absence of overt infectious virus are similar to human CNS diseases with suspected or potential viral aetiologies, such as multiple sclerosis. Therefore, MHV infection of the CNS provides a unique model in which viral replication is controlled by a vigorous immune response but the host is unable to achieve a sterile immunity, resulting in a persistent infection that is associated with ongoing pathology in the apparent absence of infectious virus.
Here, we discuss the interplay between the neurotropic viral pathogen MHV, with emphasis on the neurotropic John Howard Mueller (JHMV) strain, and the immune-mediated mechanisms that control acute and persistent CNS infection.
Mouse hepatitis virus
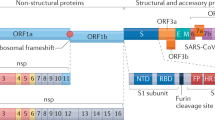
MHV is a member of the Coronaviridae family in the Order Nidovirales. The replication cycle is depicted in Fig. 1. Clinically important human coronaviruses include those that cause ∼30% of cases of the common cold and that cause severe acute respiratory syndrome (SARS)22. Bovine, porcine and avian coronaviruses also produce economically important diseases. MHV is a natural pathogen of mice that primarily infects the gastrointestinal tract. It produces a self-limiting infection with residual systemic immunological defects including reduced rejection of histo-incompatible tissues17,18. In common with many viruses, pathogenesis and immune responses depend on the viral strain, route of inoculation, age and genetic background of the host. Different MHV isolates induce various acute and chronic diseases in the murine host, including hepatitis, vasculitis, acute fatal encephalitis and encephalomyelitis associated with acute and chronic CNS demyelination17,18 (Box 2).
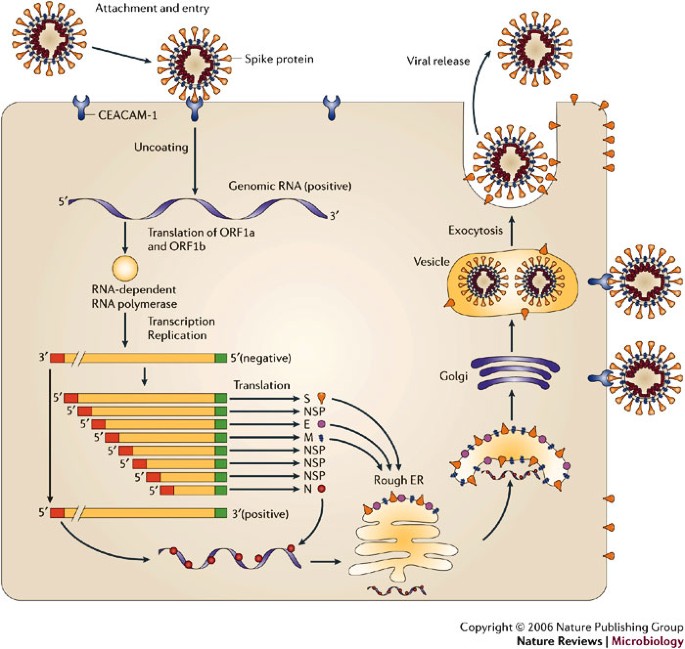
MHV binds to the host-cell receptor CEACAM-1 through interaction of the spike (S) glycoprotein. Virus entry into the host cell can occur through fusion with the surface of the host cell, with the subsequent release of the genomic RNA into the cytoplasm. Alternatively, MHV can enter the host cell through the formation of endocytic vesicles, and genomic RNA is released into the cytoplasm following fusion with the vesicle membrane (not shown). Translation of the positive-strand genomic RNA gives rise to a large polyprotein that undergoes proteolytic processing to generate an RNA-dependent RNA polymerase. Through the action of the RNA polymerase, a full-length, antisense negative-strand template is generated. Subgenomic mRNAs are synthesized, presumably from subgenomic negative-strand templates. Translation of subgenomic mRNAs gives rise to structural viral proteins. S glycoprotein is expressed on the surface of the host cell and this might contribute to fusion with neighbouring uninfected cells by binding to CEACAM-1. Virus assembly occurs within vesicles, followed by virus release by fusion of virion-containing vesicles with the plasma membrane. Released virus can infect other cells and can replicate within the parent cell through binding to CEACAM-1. E, envelope protein; ER, endoplasmic reticulum; M, membrane protein; N, nucleocapsid protein; ORF, open reading frame. Modified with permission from Ref. 22 © (2003) Macmillan Publishers Ltd.
Pathogenic strategy
MHV initiates intracellular infection by interaction of the viral-envelope spike protein (S) with its cellular receptor, the CEACAM-1 molecule23. Analysis of S genes from MHV strains that exhibit varied pathogenesis24, selection of viruses with S-gene mutations25 and recombinant viruses with modified S genes26 all confirm that the S protein is the main determinant of cell tropism and pathogenicity. But analysis of recombinant MHV that shows a high degree of tropism for neurons indicates that, in the absence of the dominant CD8+ T-cell epitope, other viral genes in addition to S genes also influence pathogenesis27,28. Adaptation to non-CEACAM-1-bearing cells can be achieved by co-culture with infected, susceptible CEACAM-1-expressing cells29, and CEACAM-1-independent infection in vitro has also been described30. These data indicate that alterations in tropism or host range might be achieved in vivo. Also, it has been suggested that receptor–S-protein affinity might contribute to the variable pathogenesis of some MHV strains31. However, not all cells that express CEACAM-1 (for instance, B cells) support MHV replication32, indicating that other (co-)receptors and intracellular factors influence productive virus replication. This is supported by the efficient replication of JHMV in the CNS, despite extremely low levels of receptor mRNA and protein expression relative to other tissues33,34. Interestingly, receptor expression on microglia decreases during CNS inflammation35, indicating that inflammatory mediators might manipulate the reservoir of susceptible cells by altering receptor expression.
Following direct intracranial injection, JHMV infection is rapidly established in the ependymal cells that line the brain ventricles36 (Fig. 2). As replication increases, virus spreads from the ependyma into the brain parenchyma. The cell types that support replication include macrophages, microglia and astrocytes, with a small number of infected oligodendrocytes in the periventricular white matter. Virus subsequently spreads down the central canal of the spinal cord, and moves out into the white matter, where it predominantly infects oligodendrocytes36. Although direct CNS injection initially disrupts BBB integrity, it is rapidly re-established and then progressively lost as inflammation increases37. Virus replication peaks at ∼5 days post infection (p.i.) but infectious virus cannot be recovered from immunocompetent hosts by ∼2 weeks p.i.16,17,18,38,39,40 (Fig. 3a). As viral titres increase, physiological changes such as alterations in BBB integrity and glial-cell activation occur in the host, even in the absence of overt clinical signs of disease. As immunity controls infectious virus, clinical signs of disease increase25,38,39,40. Infection of immunodeficient mice indicates that clinical signs are dependent on the inflammatory response, especially the CD4+ T-cell component41,42. Prolonged detection of viral antigen and mRNA in immunocompetent mice for >1 year p.i.1,16,17,18,40,43,44,45 implies that there is incomplete immunological control of CNS virus replication. A portion of the persisting viral RNAs are defective44,45, which might contribute to the failure to recover infectious virus during persistence. Virus control by inflammatory cells is associated with primary demyelination, which is ameliorated but sustained during the persistent state. Ongoing demyelination might be associated with limited virus replication and concomitant immune control.
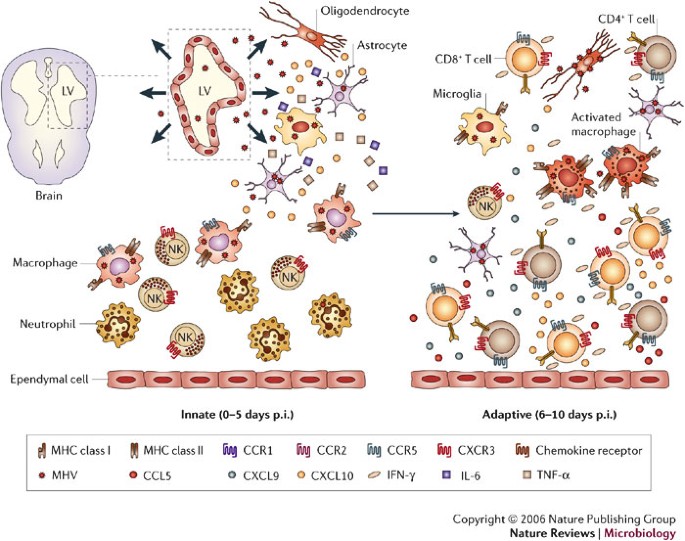
Overview of viral spread following intracranial inoculation of mouse hepatitis virus (MHV) into susceptible mice. Ependymal cells lining the lateral ventricles (LV) are the initial targets of replication, followed by spread of virus into the parenchyma and viral infection of resident glial cells of the CNS — astrocytes, oligodendrocytes and microglia. Early during acute infection, the inflammatory infiltrate consists primarily of innate components, that is, neutrophils, macrophages and natural killer (NK) cells, which presumably respond to proinflammatory signals such as TNF-α, IL-6 and CXCL10 released by glia. These proinflammatory signals enhance trafficking and accumulation of cells within the CNS. The adaptive stage of acute infection is characterized by rapid spread of virus throughout the parenchyma and increased infiltration of virus-specific CD4+ and CD8+ T cells that secrete IFN-γ, and subsequently increase expression of additional proinflammatory chemokines such as CXCL9, CXCL10 and CCL5 from astrocytes as well as inflammatory cells. Accumulation of virus-specific T cells, especially CD8+ T cells, ultimately results in a decrease in virus replication in glia. As virus replication is controlled, the number of inflammatory cells decreases, but viral persistence is associated with the retention of immune effectors in the CNS.
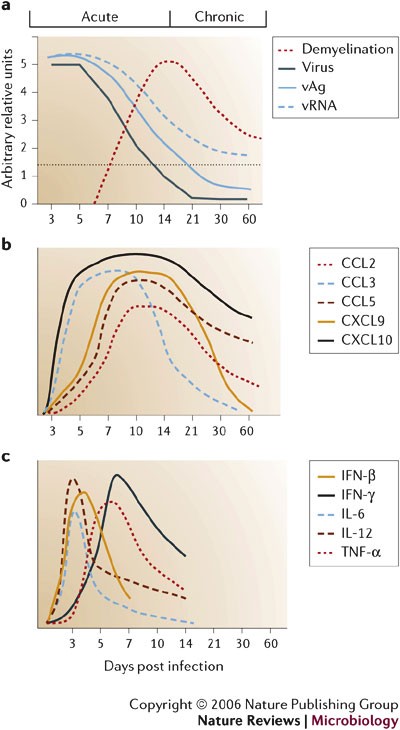
a | John Howard Mueller (JHMV) strain replication is controlled by two weeks post infection (p.i.), but viral antigen (vAg) and viral RNA (vRNA) persist. Clearance of infectious virus is accompanied by primary demyelination, which is most severe between days 14 and 21 p.i. b | Overview of the relative levels of chemokine mRNA expressed within the CNS following JHMV infection of mice. A systematic analysis of the functional contributions reveals both redundant and non-redundant roles for these molecules in participating in host defence by linking innate and adaptive immune responses (CCL3), promoting T-cell infiltration (CXCL9, CXCL10 and CCL5) and macrophage accumulation (CCL2 and CCL5). c | Schematic of cytokine mRNA kinetics during acute JHMV infection.
The innate immune response to acute infection
In common with other models of viral-induced encephalitis7,9, intranasal or direct intracranial MHV infection induces a vigorous CNS inflammatory response composed of both innate and adaptive immune components that peaks at 6–8 days p.i.37,46. CNS infection is initially manifested by rapid, dynamic and coordinated expression of chemokines, matrix metalloproteinases (MMPs), a tissue inhibitor of MMPs (TIMP-1) and pro-inflammatory cytokines (Fig. 3b,c; 0–5 days p.i.). Upregulation of these factors has largely been characterized by mRNA analysis of whole organs to reveal overall signal strength and patterns. However, more detailed analysis in a limited number of studies clearly indicates that both virus-infected and uninfected glial cells, most prominently astrocytes, provide early inflammatory signals41,47,48. Together, these molecules facilitate BBB disruption and attract innate immune effectors, which express inflammatory factors. MMP expression is associated with tissue influx of inflammatory cells, activation of cytokine secretion and CNS damage49. JHMV infection induces expression of MMP-3 mostly in astrocytes and MMP-12 mostly in oligodendrocytes, independent of the inflammatory response47,50. By contrast, a broad range of MMPs are induced in mouse models of CNS autoimmune disease49, emphasizing the distinction between CNS infection and autoimmunity as well as the complexity of CNS responses. Neutrophils, macrophages and natural killer (NK) cells are the initial inflammatory cells recruited into the MHV-infected CNS37,40 (Figs 2,4a). Secretion of pre-packaged MMP-9 by neutrophils, upregulation of adhesion molecules on CNS endothelium and, possibly, the action of IL-6 (Ref. 51) contribute to a loss of BBB integrity that facilitates the subsequent entry of further inflammatory cells into the infected CNS37. MMP-3, MMP-9 and MMP-12 mRNAs decrease either at the peak of JHMV-induced inflammation or rapidly thereafter47,50, supporting an early role in shaping the CNS environment. However, with the exception of MMP-9 (Ref. 37), their role(s) in innate inflammatory-cell trafficking and CNS pathology is unclear.
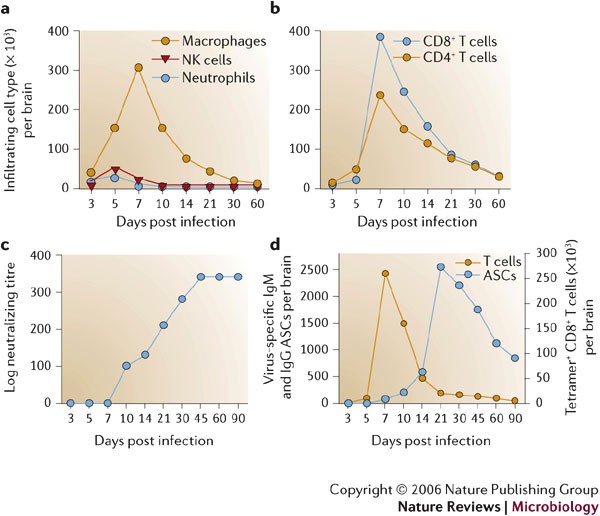
Infiltrating cells following infection of the central nervous system (CNS) with the John Howard Mueller (JHMV) strain are identified by flow cytometric analysis. Bone-marrow-derived infiltrates are distinguished from resident cells by their CD45hi phenotype and other surface markers that characterize distinct myeloid and lymphoid populations. Symbols depict representative numbers of individual cell populations within total brain cells. a–b | Macrophages make up the vast majority of early infiltrates up to day 5 following infection, whereas T cells are most abundant during peak inflammation and thereafter. c | Humoral responses emerge after infectious virus is cleared. Neutralizing antibodies in serum emerge following clearance of infectious virus and stay elevated. d | Virus-specific antibody-secreting cells (ASCs) do not emerge in the CNS until after infectious virus is cleared, and ASCs peak ∼2 weeks after maximal T-cell inflammation. Virus-specific CD8+ T cells, measured by major histocompatibility complex (MHC) class I tetramer staining, decline rapidly as virus is cleared. Compared to virus-specific CD8+ T cells, virus-specific ASCs persist at high frequencies and decline slowly, supporting a role in preventing virus recrudescence.
The earliest chemokines induced in the CNS following MHV infection are CXCL10 and CCL3 (Refs 48,52). CXCL10 is expressed by both infected and uninfected glial cells as early as day 1 p.i. (Figs 2,3b) and recruits NK cells by signalling through CXCR3 (Ref. 53). Despite rapid but transient NK-cell recruitment into the CNS, there is little direct evidence for an antiviral role; however, their potential to secrete IFN-γ might facilitate antigen presentation through upregulation of MHC class I and class II molecules. CCL3 might enhance the adaptive immune response by stimulating T-cell activation and recruitment52. Macrophages comprise the largest component of innate CNS infiltrates (Fig. 4a). Their accumulation is enhanced by CCL5 (Refs 41,54), which is induced with slightly delayed kinetics relative to CXCL10 and CCL3 (Ref. 48). Infection of the CNS with other neurotropic viruses, for example, lymphocytic choriomeningitis virus, TMEV and measles virus, induces chemokine-gene-expression profiles that are similar to MHV, which indicates that CNS-resident cells respond in a similar manner to viral infection, possibly through the expression of type I interferons (IFNs)55,56,57,58.
Cytokines that are rapidly induced by MHV, predominantly in astrocytes and microglia, include IL-1α, IL-1β, IL-6 and IL-12 (Refs 59–62) (Fig. 3c). Similar innate cytokine patterns, albeit with modified relative levels, are also characteristic of other viral CNS infections, including TMEV, vesicular stomatitis virus, HIV and West Nile virus63,64,65. This indicates that the secretion of these cytokines is a general, rather than pathogen-specific or cell-type-specific, antiviral response and is consistent with their role in the subsequent activation of adaptive immunity. TNF-α, IL-12 and IL-1β mRNA levels increase, even in the absence of inflammation59,60,62, which indicates a resident CNS cell response to MHV infection. Induction of the pleiotropic cytokine IL-6 might enhance inflammatory-cell passage across the BBB, similar to its role in the CNS autoimmune model, experimental allergic encephalomyelitis (EAE)51.
Two rapidly induced antiviral molecules, TNF-α and nitric oxide synthase-2 (iNOS, the inducible NOS isoform), which influence immunity to other CNS viral infections62,63,64,65, seem to have no role in the anti-MHV host response. Although iNOS mRNA levels increase in the CNS of MHV-infected immunocompromised mice and although iNOS suppresses virus replication in vitro66, no role for iNOS in controlling CNS virus replication was detected in vivo67,68. The reduced mortality of MHV-infected iNOS-deficient mice might be due to its contribution to neuronal apoptosis69. Despite increased TNF-α transcription during MHV infection in vitro and in vivo, translation is inhibited in MHV-infected cells70. However, TNF-α is produced by uninfected microglia within the inflamed CNS, indicating that translation might only be impaired in the minor fraction of MHV-infected microglia in vivo70. In contrast to its role as an antiviral effector and mediator of myelin loss during EAE71, neither MHV replication in vivo nor CNS pathology are altered in the absence of TNF-α70,72.
The adaptive immune response
Virus replication and spread increases despite the innate response, although innate immunity facilitates the induction, recruitment and effector function of adaptive immune components. Accumulation of virus-specific T cells, especially the CD8+ T-cell component, correlates with a marked decrease in virus replication in astrocytes, microglia, macrophages and oligodendrocytes. Distinct antiviral mechanisms control virus replication in a CNS-cell-type-specific manner. As virus replication is controlled, the number of inflammatory cells decreases; however, viral persistence is associated with the CNS retention of immune effectors.
Activation of naive T cells. Initial virus replication in the ependymal cells that line the cerebral ventricles36 (Fig. 2) probably facilitates the activation of adaptive immune responses by drainage of antigen into the cervical lymph nodes (CLN) through the cerebrospinal fluid, which connects the CNS to the lymphatic system7,8. This pathway is consistent with a model in which initial virus-specific T-cell activation occurs in the CLN, followed by chemokine-directed T-cell trafficking into the CNS. By contrast, stereotactic instillation of antigens, viruses or viral vectors directly into the CNS under conditions that maintain BBB integrity elicits poor adaptive immune responses, presumably owing to the relative isolation of the CNS and immune systems73,74. No detectable JHMV replication occurs at peripheral sites; however, virus-specific T cells are detected in the CLN prior to detection in the CNS or spleen75. Although adaptive immunity seems to be initiated in the CLN, whether infectious virus or only viral antigens are present in CLN and the identity and origin of MHV-specific antigen-presenting cells are unclear. Bone-marrow-derived circulating monocytes that are recruited into the CNS as an innate immune component might differentiate into macrophages or dendritic cells and present antigen in the CNS21,76. Alternatively, antigen-presenting cells might acquire viral antigens within the CNS and subsequently enter the CLN. The latter possibility is supported by detection of cells with a dendritic-cell-like phenotype in the CNS parenchyma and CLN as early as two days p.i.77. Therefore, it is plausible that, following phagocytosis of viral antigens and exit from the CNS, dendritic cells or macrophages in CLN provide an initial source of antigen presentation that is required for activation and expansion of virus-specific T cells.
Alterations in chemokine and cytokine patterns. Chemokine expression by infected and uninfected CNS cells and changes in receptor expression by peripherally activated adaptive immune components alter the dynamics of CNS-infiltrating cell populations. Chemokines that are expressed during the adaptive immune response to acute MHV infection include CXCL9, CXCL10, CCL2, CCL3 and CCL5, and there is corresponding expression of the chemokine receptors CCR1, CCR5 and CXCR3 (Fig. 3b) (Ref. 48). This chemokine pattern in the infected CNS is not specific for MHV infection; microglia and astrocytes synthesize chemokines following infection with both RNA and DNA viruses in the absence of inflammatory cells63,64. Similar to the innate immune response, CXCL10 is the prominent chemokine expressed during the adaptive phase of acute infection, consistent with an important role in promoting neuroinflammation. CXCL9 and CXCL10 attract activated NK and T cells that express CXCR3 (Refs 53,78,79). Supporting their central role in effector recruitment, inhibition of CXCL9 and CXCL10 increases MHV-induced mortality78,79.
Increasing accumulation of T cells as BBB integrity becomes compromised at 6–8 days p.i. coincides with a decline in neutrophils and NK cells (Fig. 4), although it is not clear if these cells exit the CNS or die in situ. By contrast, macrophages persist in the CNS; however, their phenotype alters owing to increased MHC class II expression that is driven by increasing concentrations of T-cell-derived IFN-γ. Although most early T-cell infiltrates are memory T cells specific for irrelevant antigens, these are replaced by virus-specific T cells, which expand in secondary lymphoid organs and migrate into the CNS parenchyma80. As antiviral T cells accumulate in the CNS, there is a concomitant decline in infectious virus (Fig. 3a). The reduction in CNS viral burden is reflected in the modulation of immunological markers associated with maximal viral replication. For example, chemokine transcripts that encode CXCL9, CCL2, CCL3 and CCL7 are notably reduced48. Similarly, proinflammatory cytokines (IL-1α, IL-1β, IL-6, IL-12 and IFN-β) decline60. By contrast, the T-cell chemoattractant chemokines CXCL10 and CCL5 remain elevated48, correlating with increased T-cell recruitment and IFN-γ expression40,60 (Fig. 3b,c). Unexpectedly, TNF-α mRNA levels decrease before those of IFN-γ60, although virus-specific T cells can secrete TNF-α. Among its many biological activities, IFN-γ has direct antiviral activity and induces MHC expression on CNS-resident cells, facilitating interactions between immune effectors and CNS-resident cells. In the absence of IFN-γ, MHC class I expression is reduced and MHC class II remains undetectable on microglia46,81 and most macrophages81 during JHMV infection. Indeed, peak IFN-γ mRNA levels coincide with peak T-cell infiltration, and IFN-γ protein is functionally evident in the inflamed CNS by maximal expression of both MHC class I and II on microglia46,60,81.
T-cell infiltration and antiviral effector functions. Novel concepts emerging from MHV-induced CNS infection are the differential abilities of T-cell subsets to migrate within the CNS and the crosstalk between T-cell subsets. CD4+ T cells cross the BBB, but instead of trafficking to parenchymal sites of virus replication, they accumulate around blood vessels82. By contrast, CD8+ T cells enter the parenchyma after migrating through the BBB. The differential ability of CD4+ T cells versus CD8+ T cells to traffic through the infected tissue is associated with expression of TIMP-1 by CD4+ T cells but not CD8+ T cells47. These data indicate that, rather than expression of a protease to promote migration, expression of a protease inhibitor prevents migration of CD4+ T cells into the CNS parenchyma. In the absence of CD4+ T cells, parenchymal CD8+ T-cell infiltration is dramatically decreased and is associated with increased apoptosis82, indicating that CD4+ T cells, either directly or indirectly, provide factors that are required for both the migration and the survival of CD8+ T cells within the CNS. Although IL-2 has been excluded, other survival factors remain unidentified83.
During peak T-cell accumulation, most CD8+ and CD4+ T cells within the CNS are virus specific16,40. Virus-specific CD8+ T cells accumulate to 10-fold higher frequencies in the CNS compared with the periphery and they express the CD44hi, CD62L−/lo, CD11ahi and CD49d (VLA-4) activation/memory phenotypic markers40, which is consistent with their crucial role in controlling acute MHV replication40,75. CD43hi and CD127−/lo expression discriminates virus-specific CD8+ T cells within the CNS from those T cells specific for irrelevant antigens, which retain a CD43int, CD127+ phenotype80. Although the early activation marker CD69 is only transiently upregulated early during priming and expansion of T cells in secondary lymphoid organs, CD8+ T cells recruited into the CNS during JHMV infection retain CD69 expression40, consistent with other CNS-inflammation models84.
Virus-specific CD8+ T cells isolated from the acutely inflamed CNS secrete IFN-γ, express granzyme B and are efficient cytolytic effectors40,85. These T cells accumulate within the CNS coincident with inhibition of infectious virus, and transferred memory CD8+ T cells control virus replication in immunodeficient hosts40,46, confirming their role as primary effectors of virus clearance. Compared with highly activated CD8+ T cells obtained during acute infection, virus-specific memory T cells are superior at controlling virus replication in immunodeficient hosts42,72. This enigma might reflect an increased sensitivity of highly activated CD8+ T cells to activation-induced apoptosis, or their preferential accumulation in peripheral compartments86.
T-cell antiviral effector mechanisms are cell-type specific. In mice deficient in perforin-mediated cytolysis, viral replication is uncontrolled in macrophages, microglia and astrocytes38. However, infection of oligodendrocytes is controlled in the absence of cytolysis38. These results indicate that an effector mechanism distinct from MHC class I recognition by CD8+ T cells controls virus replication in oligodendrocytes. By contrast, the absence of the Fas/FasL cytolytic pathway does not alter pathogenesis, virus clearance or pathology87. In IFN-γ-deficient mice that are competent for perforin-mediated cytolysis, virus replication is controlled in astrocytes and microglia, but not oligodendrocytes39. The distinct use of effector mechanisms in the control of viral replication by CD8+ T cells was confirmed by adoptive transfer of CD8+ T cells deficient in either cytolytic activity or IFN-γ secretion into infected immunodeficient hosts40,46. Furthermore, infection of mice with a selective defect in IFN-γ signalling in oligodendrocytes confirms that direct IFN-γ signalling is required to control oligodendrocyte infection88. These data support the concept that the mechanisms of CD8+ T-cell-dependent control of virus replication are cell-type dependent.
Pathway to persistent infection
After infectious MHV is eliminated at ∼2 weeks p.i., inflammatory cells, viral antigen and viral mRNA persist in the CNS (Fig. 3a). Virus-specific CD8+ T-cell cytolytic activity is rapidly lost by day 14 p.i., as viral-antigen concentrations decrease40,85. Whether the loss of cytolytic function is due to decreased antigen89 or reflects an attempt to limit the potential adverse effects of cytolysis on CNS cells is not clear.
The contribution of CD8+ T-cell escape variants to persistent infection depends on mouse strain, age and immune status. Little evidence for escape mutants has been detected during virus persistence in naive mice infected as adults45 or in mice undergoing reactivation owing to the absence of humoral immunity38. Nevertheless, progressive accumulation of viral quasispecies with deletions in the S-protein hypervariable domain, which contains the immunodominant H-2b CD8+ T-cell epitope, was found in persistently infected H-2b mice. Secondary-structure analysis indicated that the deleted regions reside in an RNA stem-loop structure that forms a 'hot spot' for RNA recombination90, questioning the extent to which the S mutants emerged from immune pressure. Mutations in this S-protein epitope were clearly associated with increased infectious virus in the CNS following infection of neonatal mice protected by maternal antibody91. A potential for immune escape was also shown when pre-immune mice that harboured CD8+ T cells specific for a novel epitope were challenged with the recombinant MHV-A59 strain that expressed the same epitope27. Taken together, these data indicate that T-cell escape variants do not have a prominent role in the persistence of virus after infection of naive adult mice, but might readily emerge in genome regions that do not affect viral fitness, especially under conditions of pre-existing antibody or T-cell memory.
CD8+ T cells that are found in persistent CNS MHV infection are not impaired in IFN-γ secretion, which indicates that loss of cytolytic function is not due to the induction of an anergic state. However, impaired virus-induced TNF-α secretion by CD8+ T cells during both acute infection and persistence85 indicates that T-cell retention within the CNS might be due to decreased secretion of apoptosis-inducing factors. The loss of CD8+ T-cell-mediated cytolysis during resolution of primary MHV infection and throughout persistence contrasts with the retention of cytolytic effector function in reactivated T-memory cells following neurotropic influenza-virus challenge84. However, increased granzyme B levels in reactivated MHV-specific memory CD8+ T cells, compared with primary CD8+ T cells isolated from the CNS following challenge, supported the retention of intrinsic cytolytic function85. These data show that the loss of virus-specific cytolytic function is not an intrinsic property of the inflamed CNS environment, but reflects distinct differentiation states of primary CD8+ T cells compared with vaccine-induced memory CD8+ T cells.
Virus-specific T cells decline markedly between 10 and 21 days p.i., but are retained for at least 3 months following clearance of infectious virus43,85. The initial T-cell decline in the CNS is similar to, but not as prominent as, the decline of T-cell effector populations in peripheral lymphoid organs following antigen elimination and withdrawal of cytokine survival factors92,93. Nevertheless, CNS retention of small numbers of both CD4+ and CD8+ T cells40,43 indicates that the myelin-loss characteristic of the persistent phase of MHV infection is associated with a continuing immune response, sustained by low-level oligodendrocyte infection. Sustained CD69 expression also distinguishes CD8+ T cells that are retained within the CNS from resting peripheral memory cells in lymphoid organs, and suggests chronic activation40 or an effector memory phenotype characteristic of memory T cells residing in non-lymphoid tissues92. Antigen-driven T-cell persistence was indicated by the limited T-cell-receptor specificities found in CD8+ T-cell populations isolated during MHV persistence compared with T cells isolated during acute infection94. Complete disappearance of both CD8+ and CD4+ T cells from the CNS following infection with a neurotropic MHV43 not associated with viral persistence or myelin loss25 supports a role for viral persistence or continuing pathology in maintaining T-cell retention.
The contribution of local proliferation or ongoing recruitment to the T-cell population that persists in the CNS remains unclear. Indeed, IL-15, which regulates antigen-independent homeostasis of memory cells in lymphoid organs93, is not required for CD8+ T-cell retention in the CNS (C.C.B., unpublished data). Adoptive transfer of CD8+ T cells into persistently infected mice further indicates that there is limited recruitment to the CNS compared with the acute phase (C.C.B., unpublished data). These data are consistent with the recent observations that memory T cells traffic poorly into the CNS92 and that activated T cells recruited in response to acute infection are only retained within the CNS on cognate antigen recognition2,7,8. Overall, analysis of persistent MHV infection indicates that CD8+ T-cell turnover within the CNS is limited and does not comprise significant ongoing peripheral recruitment.
Humoral effectors and control of CNS persistence
Serum antibody that is present prior to MHV infection, either due to systemic administration or immunization, provides protection, although not necessarily by inhibition of virus replication. Virus-neutralizing antibody and antibodies with no apparent neutralizing activity modify MHV-induced CNS disease if passively transferred prior to infection17,18. Transport of neutralizing antibody into the CNS parenchyma owing to the loss of BBB integrity50 might limit the replication of challenge virus by inhibiting receptor binding. A complement-dependent role in protection for antibodies lacking neutralizing activity is less clear95 although at least one nucleocapsid-protein-derived epitope is expressed on the MHV-infected cell surface96, providing a potential recognition structure.
Antibody responses in infected naive animals are delayed relative to the vigorous cell-mediated immune response (Fig. 4c,d). Serum antibody, including neutralizing antibody, is virtually undetectable and predominantly limited to IgM prior to the complete elimination of infectious virus (Fig. 4c,d). Furthermore, mice that lack humoral immunity control CNS-infectious virus with kinetics similar to immunocompetent mice, accompanied by a normal inflammatory response during acute infection97,98. These data are consistent with the concept that control of acute infection is independent of humoral immunity17,18. However, in contrast to wild-type mice that recover, mice that are unable to secrete antibody show increased mortality after resolution of acute disease, associated with the re-emergence of infectious virus within the CNS97,98. Interestingly, the A59 strain of MHV, which infects both the liver and CNS, fails to reactivate in the liver in the absence of humoral immunity99. Whether this is due to the absence of viral persistence in liver or reflects a fundamental difference in immune control in these two organs is not clear. Passive transfer of neutralizing, but not nonneutralizing, viral-specific antibody into B-cell-deficient mice following initial virus clearance prevents virus reactivation, confirming the crucial role of antibody in regulating CNS viral persistence100. The inability of transferred non-neutralizing antibody to prevent virus recrudescence is inconsistent with the apparent protective role for non-neutralizing antibody prior to infection. Interestingly, infectious virus reactivates as passive antibody levels decline, supporting a requirement for CNS retention of antibody-secreting cells (ASCs) in providing long-term control of persistence.
MHV-specific ASCs accumulate rapidly after control of infectious virus during persistence101 (Figs 4c,5). Although ASCs that are not specific for MHV are present in the CNS during the virus-clearance phase, only a few virus-specific ASCs are detectable in either the CNS or peripheral lymphoid system during acute infection101, consistent with the inability to detect serum antibody. Both populations are retained after virus is cleared101. The preceding peak of virus-specific IgG ASCs in CLN ∼1 week prior to peak CNS accumulation indicates initial ASC activation and differentiation in CLN and spleen prior to CNS migration. Virus-specific ASCs are retained in the CNS at high frequencies for at least 3 months p.i., indicating that ASC-specific survival factors are present in the CNS during viral persistence. Despite their progressive decline, virus-specific ASCs are maintained at higher levels than virus-specific T cells40,101. The CNS has been shown to be a survival niche for ASCs following Sindbis-virus- and Semliki-Forest-virus-induced encephalitis102,103. The accumulation and maintenance of virus-specific ASCs in the CNS, coupled with reactivation of infectious virus in the absence of antibody, indicates that antibody secretion within the CNS, and not T-cell immunity, is crucial for the control of MHV CNS persistence.
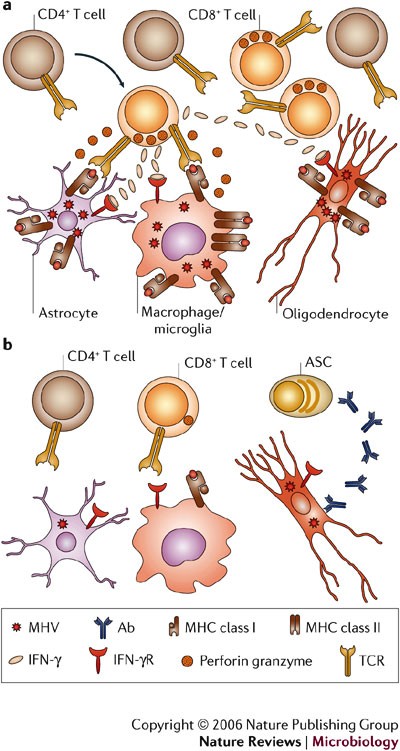
a | CD8+ T cells are crucial for elimination of replicating virus during acute infection. The direct antiviral role of CD4+ T cells is unclear; however, they enhance CD8+ T-cell survival and function by an unknown mechanism. Whereas perforin-mediated mechanisms, in the absence of IFN-γ, control virus in astrocytes and microglia, IFN-γ is crucial for reducing infection in oligodendrocytes. Major histocompatibility complex (MHC) class I expression on astrocytes is postulated, as perforin-mediated cytolysis requires class I expression; however, this has not been demonstrated in vivo. Increased IFN-γ release by T cells during interaction with virus-infected targets enhances class I expression on all glial cells and induces class II expression on microglia, therefore further enhancing target–T-cell interactions. b | As viral antigen is cleared, CD8+ T cells lose cytolytic function and virus persists predominantly in oligodendrocytes. IFN-γ secretion decreases, MHC is downregulated and T cells decline but persist at low frequencies. Virus-specific antibody-secreting cells are crucial to prevent virus recrudescence. Ab, antibody; IFN-γR, IFN-γ receptor; MHV, mouse hepatitis virus; TCR, T-cell receptor.
Conclusions and future perspectives
Analysis of the MHV model highlights the diversity of immune responses that is required to prevent subsequent pathology associated with a persistent infection confined to a single target organ. This model supports a paradigm in which cell-mediated immunity affects clearance of infectious virus through mechanisms that are dictated by the specific cell type within the infected tissue (Fig. 5). Although effective in controlling acute virus replication, T cells are ultimately unable to achieve sterile immunity or suppress virus reactivation, most likely owing to downregulation or inhibition of destructive effector functions in vivo. However, cessation of T-cell function is complemented by a wave of virus-specific ASCs that are recruited into the CNS following resolution of acute infection. In contrast to T cells, ASCs are maintained within the CNS at high frequencies during virus persistence. These data indicate that local secretion of neutralizing antibody within the CNS maintains virus at low levels, thereby providing a protective in situ effector system preventing virus recrudescence (Fig. 5).
Many issues related to neurotropic MHV infections remain unresolved. Contributions of alternative receptors, co-receptors or receptor-independent spread to tropism and pathogenesis are still elusive. Viral components involved in cell signalling through viral receptors, Toll-like receptors or type I IFN pathways are also largely unexplored. Distinct MHV isolates, combined with powerful new genetic tools26,27,28, promise to shed light on these pathways. Differential cell susceptibility to antiviral mechanisms also requires further investigation38,39,46. Specifically, the ability of mature glial cells to present viral antigens104, regulation of ligands affecting lymphocyte function, and factors involved in apoptosis are of interest. Similarly, the responsiveness of resident CNS cells to IFNs in vivo is largely unknown88. The role of dendritic cells during virus-induced CNS inflammation, as well as CD4+ T-cell contributions to CD8+ T-cell function within the CNS82, also requires evaluation. Last, an intriguing question is how, and in what form, virus persists, although a replication-competent form is implicated by virus recrudescence in the absence of humoral immunity97,98. Resolving mechanisms of viral persistence might also elucidate events associated with ongoing immune activation and T-cell and ASC retention, all potentially contributing to demyelinating disease.
Acute, potentially lethal viral infections of the human CNS, for example, West Nile virus and Saint Louis encephalitis virus, primarily target neurons105. Other human viruses, for example, herpes viruses, target and remain latent in neurons. HIV and JC polyomavirus primarily target other CNS cell types and are prone to producing latent or persistent CNS infections9,12,13. Although it is unclear how SARS-virus CNS replication contributes to pathogenesis, recent data also confirm CNS virus infection106.
Coronavirus infection of the CNS has provided unique insights into the immune regulation of acute and persistent infection at the cellular level of a natural rodent pathogen, and provides a model for studying chronic demyelinating diseases, such as multiple sclerosis. Delineation of the dynamic interactions that regulate acute and persistent infections of the CNS has implications for vaccine design as well as for the development of novel immunotherapeutics to limit viral replication and attenuate the potential damaging effects of the immune response within the CNS.
References
- Fabry, Z., Raine, C. S. & Hart, M. N. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol. Today 15, 218–224 (1994).
Hickey, W. F. Basic principles of immunological surveillance of the normal central nervous system. Glia 36, 118–124 (2001).
Aloisi, F., Ria, F. & Adorini, L. Regulation of T-cell responses by CNS antigen-presenting cells: different roles for microglia and astrocytes. Immunol. Today 21, 141–147 (2000).
Johnson, M. D., Gold, L. I. & Moses, H. L. Evidence for transforming growth factor-β expression in human leptomeningeal cells and transforming growth factor-β-like activity in human cerebrospinal fluid. Lab. Invest. 67, 360–368 (1992).
Hoek, R. M. et al. Down-regulation of the macrophage lineage through interaction with OX2 (CD200). Science 290, 1768–1771 (2000).
Neumann, H., Misgeld, T., Matsumuro, K. & Wekerle, H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc. Natl Acad. Sci. USA 95, 5779–5784 (1998).
Dorries, R. The role of T-cell-mediated mechanisms in virus infections of the nervous system. Curr. Top. Microbiol. Immunol. 253, 219–245 (2001).
Ransohoff, R. M., Kivisakk, P. & Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nature Rev. Immunol. 3, 569–581 (2003).
Griffin, D. E. Immune responses to RNA-virus infections of the CNS. Nature Rev. Immunol. 3, 493–502 (2003).
Belkaid, Y. & Rouse, B. T. Natural regulatory T cells in infectious disease. Nature Immunol. 6, 353–360 (2005).
Jiang, H. & Chess, L. An integrated view of suppressor T cell subsets in immunoregulation. J. Clin. Invest. 114, 1198–1208 (2004).
Lipton, H. & Gilden, D. Viral diseases of the central nervous system: persistent infections. In Viral Pathogenesis (ed. Nathanson, N.) 855–870 (Lippincott–Rave, Philadelphia, 1997).
Rall, G. & Oldstone, M. Viral persistence in the central nervous system. In In Defense Of The Brain: Current Concepts In The Immunopathogenesis And Clinical Aspects Of CNS Infection 273–289 (Blackwell Science, Malden, 1997).
Fazakerley, J. & Walker, R. Virus demyelination. J. Neurovirol. 9, 148–164 (2003).
Tsunoda, I. & Fujinami, R. Theiler's murine encephalomyelitis virus. In Persistent Viral Infections (eds Ahmed, R. & Chen, I.) 517–536 (John Wiley, New York, 1999).
Marten, N. W., Stohlman, S. A. & Bergmann, C. C. MHV infection of the CNS: mechanisms of immune-mediated control. Viral Immunol. 14, 1–18 (2001).
Stohlman, S., Bergmann, C. & Perlman, S. Mouse hepatitis virus. In Persistent Viral Infection (eds Ahmed, R. & Chen, I.) 537–558 (John Wiley, New York, 1999).
Perlman, S. Pathogenesis of coronavirus-induced infections: review of pathological and immunological aspects. Adv. Exp. Med. Biol. 440, 503–513 (1998).
Trotter, M., Schlitt, B. P., Kung, A. Y. & Lipton, H. L. Transition from acute to persistent Theiler's virus infection requires active viral replication that drives proinflammatory cytokine expression and chronic demyelinating disease. J. Virol. 78, 12480–12488 (2004).
Croxford, J. L., Olson, J. K. & Miller, S. D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler's virus-induced demyelinating disease model of multiple sclerosis. Autoimmun. Rev. 1, 251–260 (2002).
McMahon, E. J., Bailey, S. L., Castenada, C. V., Waldner, H. & Miller, S. D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nature Med. 11, 252–253 (2005).
Stadler, K. et al. SARS — beginning to understand a new virus. Nature Rev. Microbiol. 1, 209–218 (2003).
Williams, R. K., Jiang, G. S., Snyder, S. V., Frana, M. F. & Holmes, K. V. Purification of the 110-dilodalton glycoprotein receptor for mouse hepatitis virus (MHV)-A59 and identification of a non-functional, homologous protein MHV-resistant SJL/J mice. J. Virol. 64, 3817–3823 (1990). This paper is the first to describe the host-cell receptor for murine coronaviruses.
Parker, S. E., Gallagher, T. M. & Buchmeier, M. J. Sequence analysis reveals extensive polymorphism and evidence of deletions within the E2 glycoprotein gene of several strains of murine hepatitis virus. Virology 173, 664–673 (1989). This study was the first to clearly identify a polymorphic region of the S protein in different strains of MHV. These observations provided the potential for understanding viral determinants of pathogenicity at the molecular level.
Fleming, J. O., Trousdale, M., El-Zaatari, F., Stohlman, S. A. & Weiner, L. P. Pathogenicity of antigenic variants of murine coronavirus JHM selected with monoclonal antibodies. J. Virol. 58, 869–875 (1986).
Phillips, J. J., Chua, M. M., Lavi, E. & Weiss, S. R. Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J. Virol. 73, 7752–7760 (1999). Recombinant technology that replaced the viral S proteins and analysis of subsequent pathogenesis showed that the S protein is a major determinant of coronavirus-induced CNS disease.
Chua, M., MacNamara, K., Mateo, L., Shen, H. & Weiss, S. Effects of an epitope specific CD8+T cell response on murine central nervous system disease: protection from virus replication and antigen spread and selection of epitope escape mutants. J. Virol. 78, 1150–1159 (2004).
MacNamara, K., Chua, M., Phillips, J. & Weiss, S. Contributions of viral genetic background and a single amino acid substitution in an immunodominant CD8+ T cell epitope to murine coronavirus neurovirulence. J. Virol. 79, 9108–9118 (2005).
Chen, W. & Baric, R. S. Molecular anatomy of mouse hepatitis virus persistence: coevolution of increased host cell resistance and virus virulence. J. Virol. 70, 3947–3960 (1996).
Gallagher, T. M., Buchmeier, M. J. & Perlman, S. Cell receptor-independent infection by a neurotropic murine coronavirus. Virology 191, 517–522 (1992).
Gallagher, T. & Buchmeier, M. Coronavirus spike proteins in viral entry and pathogenesis. Virology 279, 371–374 (2001).
Morales, S., Parra, B., Ramakrishna, C., Blau, D. & Stohlman, S. B cell mediated lysis of cells infected with the neurotropic JHM strain of mouse hepatitis virus. Virology 286, 160–167 (2001).
Nakagaki, K., Nakagaki, K. & Taguchi, F. Receptor-independent spread of a high neurotropic murine coronavirus JHMV strain from initially infected microglial cells in mixed neural cultures. J. Virol. 79, 6102–6110 (2005).
Ontiveros, E., Kim, T. S., Gallagher, T. M. & Perlman, S. Enhanced virulence mediated by the murine coronavirus, mouse hepatitis strain JHM, is associated with a glycine at residue 310 of the spike glycoprotein. J. Virol. 77, 10260–10269 (2003).
Ramakrishna, C., Bergmann, C., Holmes, K. V. & Stohlman, S. Expression of the mouse hepatitis virus receptor by central nervous system microglia. J. Virol. 78, 7828–7832 (2004).
Wang, F. I., Hinton, D. R., Gilmore W., Trousdale, M. D. & Fleming, J. O. Sequential infection of glial cells by the murine hepatitis virus JHM strain (MHV-4) leads to a characteristic distribution of demyelination. Lab. Invest. 66, 744–754 (1992).
Zhou, J., Stohlman, S., Hinton, D. R. & Marten, N. Neutrophils modulate inflammation during viral induced encephalitis. J. Immunol. 170, 3331–3336 (2003).
Lin, M., Stohlman, S. & Hinton, D. Mouse hepatitis virus is cleared from the central nervous system of mice lacking perforin-mediated cytolysis. J. Virol. 71, 383–391 (1997). The authors provide the initial evidence that perforin-mediated cytolysis controls virus replication in only a subset of CNS cells during acute infection.
Parra, B. et al. g interferon is required for viral clearance from central nervous system oligodendroglia. J. Immunol. 162, 1641–1647 (1999). This report shows the crucial role of IFN-γ in coronavirus-induced CNS disease and provides the first evidence that separate effector mechanisms control virus replication within a single target, the CNS.
Bergmann, C., Altman, J., Hinton, D. & Stohlman, S. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the CNS. J. Immunol. 163, 3379–3387 (1999). One of the first reports of the application of tetramer technology to an infectious disease. The data quantify both recruitment and retention of virus-specific CD8+ T cells in the CNS.
Lane, T. E. et al. A central role for CD4+ T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J. Virol. 74, 1415–1424 (2000).
Wu, G. F., Dandekar, A. A., Pewe, L. & Perlman, S. CD4 and CD8 T cells have redundant but not identical roles in virus-induced demyelination. J. Immunol. 165, 2278–2286 (2000).
Marten, N., Stohlman, S. & Bergmann, C. Role of viral persistence in retaining CD8+ T cells within the central nervous system. J. Virol. 74, 7903–7910 (2000).
Adami, C. et al. Evolution of mouse hepatitis virus (MHV) during chronic infection: quasispecies nature of the persisting MHV RNA. Virology 209, 337–346 (1995).
Bergmann, C. C. et al. Variability of persisting MHV RNA sequences constituting immune and replication relevant domains. Virology 244, 563–572 (1998).
Bergmann, C. et al. Perforin mediated effector function within the CNS requires IFN-γ mediated MHC upregulation. J. Immunol. 170, 3204–3213 (2003).
Zhou, J. et al. Expression of matrix metalloproteinases and their tissue inhibitor during viral encephalitis. J. Virol. 79, 4764–4773 (2005).
Lane, T. E. et al. Dynamic regulation of α- and β-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J. Immunol. 160, 970–978 (1998).
Yong, V. W., Power, C., Forsyth, P. & Edwards, D. R. Metalloproteinases in biology and pathology of the nervous system. Nature Rev. Neuroscience 2, 502–511 (2001).
Zhou, J., Stohlman, S., Atkinson, R., Hinton, D. & Marten, N. Matrix metalloproteinase expression correlates with virulence following neurotropic mouse hepatitis virus infection. J. Virol. 76, 7373–7384 (2002).
Ishihara, K. & Hirano, T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 13, 357–368 (2002).
Trifilo, M. J., Bergmann, C. C., Kuziel, W. A. & Lane, T. E. CC chemokine ligand 3 (CCL3) regulates CD8+-T-cell effector function and migration following viral infection. J. Virol. 77, 4004–4014 (2003).
Trifilo, M. J. et al. CXC chemokine ligand 10 controls viral infection in the central nervous system: evidence for a role in innate immune response through recruitment and activation of natural killer cells. J. Virol. 78, 585–594 (2004).
Glass, W. G. et al. Antibody targeting of the CC chemokine ligand 5 (CCL5) results in diminished leukocyte infiltration into the central nervous system and reduced neurologic disease in a viral model of multiple sclerosis. J. Immunol. 172, 4018–4025 (2004).
Asensio, V. C. & Campbell, I. L. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J. Virol. 71, 7832–7840 (1997).
Hoffman, L. M., Fife, B. T., Begolka, W. S., Miller, S. D. & Karpus, W. J. Central nervous system chemokine expression during Theiler's virus-induced demyelinating disease. J. Neurovirol. 5, 635–642 (1999).
Manchester, M., Eto, D. S. & Oldstone, M. B. Characterization of the inflammatory response during acute measles encephalitis in NSE-CD46 transgenic mice. J. Neuroimmunol. 96, 207–217 (1999).
Salazar-Mather, T. P., Lewis, C. A. & Biron, C. A. Type I interferons regulate inflammatory cell trafficking and macrophage inflammatory protein 1α delivery to the liver. J. Clin. Invest. 110, 321–330 (2002).
Pearce, B. D., Hobbs, M. V., McGraw, T. S. & Buchmeier, M. J. Cytokine induction during T-cell-mediated clearance of mouse hepatitis virus from neurons in vivo. J. Virol. 68, 5483–5495 (2004).
Parra, B., Hinton, D. R., Lin, M. T., Cua, D. J. & Stohlman, S. A. Kinetics of cytokine mRNA expression in the CNS following lethal and sublethal coronavirus-induced encephalomyelitis. Virology 233, 260–270 (1997).
Rempel, J. D., Murray, S. J., Meisner, J. & Buchmeier. M. J. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 318, 381–392 (2004).
Rempel, J. D., Quina, L. A., Blakelu-Gonzales, P. K., Buchmeier, M. J. & Gruol, D. L. Viral induction of central nervous system innate immune responses. J. Virol. 79, 4369–4381 (2005).
Lane, T. E. & Buchmeier, M. J. Chemokine responses in virus-induced neurologic disease: balancing host defense and neuropathology. In Universe In Delicate Balance: Chemokines And The Nervous System (ed. Ransohoff, R.) 191–202 (Elsevier, Amsterdam, 2002).
Asensio, V. C. & Campbell, I. L. Chemokines and viral diseases of the central nervous system. Adv. Virus Res. 56, 127–173 (2001).
Biron, C. A. Role of early cytokines, including α and β interferons (IFN-α/β), in innate and adaptive immune responses to viral infections. Semin. Immunol. 10, 383–390 (1998).
Lane, T. E., Paoletti, A. D. & Buchmeier, M. J. Disassociation between the in vitro and in vivo effects of nitric oxide on a neurotropic murine coronavirus. J. Virol. 71, 2202–2210 (1997).
Lane, T. E., Fox, H. S. & Buchmeier, M. J. Inhibition of nitric oxide synthase-2 reduces the severity of mouse hepatitis virus-induced demyelination: implications for NOS2/NO regulation of chemokine expression and inflammation. J. Neurovirol. 5, 48–54 (1999).
Wu, G. F., Pewe, L. & Perlman, S. Coronavirus-induced demyelination occurs in the absence of inducible nitric oxide synthase. J. Virol. 74, 7683–7686 (2000).
Chen, B. P. & Lane, T. E. Lack of nitric oxide synthase type 2 (NOS2) results in reduced neuronal apoptosis and mortality following mouse hepatitis virus infection of the central nervous system. J. Neurovirol. 8, 58–63 (2002).
Stohlman, S. A. et al. Tumor necrosis factor expression during mouse hepatitis virus induced demyelination. J. Virol. 69, 5898–5903 (1995).
Sacca, R., Cuff, C. A. & Ruddle, N. H. Mediators of inflammation. Curr. Opin. Immunol. 9, 851–857 (1997).
Pewe, L. & Perlman, S. Cutting edge: CD8 T cell-mediated demyelination is IFN-γ dependent in mice infected with a neurotropic coronavirus. J. Immunol. 168, 1547–1551 (2002).
Stevenson, P. G., Hawke, S., Sloan, D. J. & Bangham, C. R. M. The immunogenicity of intracerebral virus-infection depends on anatomical site. J. Virol. 71, 145–151 (1997).
Lowenstein, P. R. Immunology of viral-vector-mediated gene transfer into the brain: an evolutionary and developmental perspective. Trends Immunol. 23, 23–30 (2002).
Marten, N., Stohlman, S., Zhou, Z. & Bergmann, C. Kinetics of virus specific CD8+T cell expansion and trafficking following central nervous system infection. J. Virol. 77, 2775–2778 (2003).
Greter, M. et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nature Med. 11, 328–334 (2005).
Trifilo, J. J. & Lane, T. E. The CC chemokine ligand 3 regulates CD11c+CD11b+CD8α-dendritic cell maturation and activation following viral infection of the central nervous system: implications or a role in T cell activation. Virology 327, 8–15 (2004).
Liu, M. T. et al. Cutting edge: the T cell chemoattractant IFN-inducible protein 10 (IP-10) is essential in host defense against viral-induced neurologic disease. J. Immunol. 165, 2327–2330 (2000). This paper was the first to show that expression of CXCL10 within the CNS in response to viral infection was crucial in host defence by attracting virus-specific T cells.
Liu, M. T., Armstrong, D., Hamilton, D. A. & Lane, T. E. Expressing of Mig (monokine induced by interferon γ) is important in T lymphocyte recruitment and host defense following viral infection of the central nervous system. J. Immunol. 166, 1790–1795 (2001).
Chen, A. M., Khanna, N., Stohlman, S. A. & Bergmann, C. C. Virus-specific and bystander CD8 T cells recruited during virus-induced encephalomyelitis. J. Virol. 79, 4700–4708 (2005).
Bergmann, C. et al. Perforin and interferon γ mediated control of coronavirus central nervous system infection by CD8 T cells in the absence of CD4 T cells. J. Virol. 78, 1739–1750 (2004).
Stohlman, S. A., Bergmann, C. C., Lin, M. T., Cua, D. J. & Hinton, D. R. CTL effector function within the CNS requires CD4+ T cells. J. Immunol. 160, 2896–2904 (1998).
Zhou, J., Hinton, D. R., Stohlman, S. A. & Marten, N. Maintenance of CD8+ T cells during acute viral infection of the central nervous system requires CD4+ T cells but not interleukin-2. Virol. Immunol. 18, 162–169 (2005).
Hawke, S., Stevenson, P. G., Freeman, S. & Bangham, C. R. M. Long term persistence of activated cytotoxic T lymphocytes after viral infection of the central nervous system. J. Exp. Med. 187, 1575–1582 (1998). The authors show that activated virus-specific CD8+ T cells derived from a memory population are retained within the CNS in the absence of viral antigen. Antigen-specific T cells can persist and retain effector function within tissues of relative immune privilege, and might be important for efficient control of viral recrudescence.
Ramakrishna, C., Stohlman, S., Atkinson, R., Hinton, D. H. & Bergmann, C. C. Differential regulation of primary and secondary CD8+ T cells in the CNS. J. Immunol. 173, 6265–6273 (2004).
John, B. & Crispe, I. N. Passive and active mechanisms trap activated CD8+ T cells in the liver. J. Immunol. 172, 5222–5229 (2004).
Parra, B. et al. Contributions of Fas–Fas ligand interactions to the pathogenesis of mouse hepatitis virus in the central nervous system. J. Virol. 74, 2447–2450 (2000).
Gonzalez, J. M. et al. Expression of a dominant negative IFN-γ receptor on mouse oligodendrocytes. Glia 51, 22–34 (2005).
Harty, J. T., Tvinnereim, A. R. & White, D. W. CD8+ T cell effector mechanisms in resistance to infection. Annu. Rev. Immunol. 18, 275–308 (2000).
Rowe, C. L. et al. Generation of Coronavirus spike deletion variants by high frequency recombination at regions of predicted RNA secondary structure. J Virol. 71, 6183–6190 (1997).
Pewe, L., Wu, G. F., Barnett, E. M., Castro, R. F. & Perlman, S. Cytotoxic T cell-resistant variants are selected in a virus-induced demyelinating disease. Immunity 5, 253–262 (1996). These authors provide the first evidence that CD8+ T-cell escape mutants that evade the developing immune system are the basis for coronavirus reactivation within the CNS of mice infected as neonates and protected by maternal antibody.
Lefrancois, L. & Masopust, D. T cell immunity in lymphoid and non-lymphoid tissues. Curr. Opin. Immunol. 14, 503–508 (2002).
Masopust, D. & Ahmed, R. Reflections on CD8 T-cell activation and memory. Immunol. Res. 29, 151–160 (2004).
Marten, N. et al. Selection of CD8+ T cells with highly focused specificity during viral persistence in the central nervous system. J. Immunol. 162, 3905–3914 (1999).
Fleming, J. O., Shubin, R. A., Sussman, M. A., Casteel, N. & Stohlman, S. A. Monoclonal antibodies to the matrix (E1) glycoprotein of mouse hepatitis virus protect mice from encephalitis. Virology 168, 162–167 (1988).
Kyuwa, S., Cohen, M., Nelson, G., Tahara, S. & Stohlman, S. A. Modulation of macromolecular synthesis by coronavirus: implications for pathogenicity. J. Virol. 68, 6815–6819 (1994).
Lin, M. T., Hinton, D. R., Marten, N. W., Bergmann, C. C. & Stohlman, S. A. Antibody prevents virus reactivation within the central nervous system. J. Immunol. 162, 7358–7368 (1999).
Ramakrishna, C., Stohlman, S., Atkinson, R., Schlomchik, M. & Bergmann, C. Mechanisms of central nervous system viral persistence: critical role of antibody and B cells. J. Immunol. 168, 1204–1211 (2002).
Matthews, A. E. et al. Antibody is required for clearance of infectious murine hepatitis virus A59 from the central nervous system, but not the liver. J. Immunol. 167, 5254–5263 (2001).
Ramakrishna, C., Bergmann, C., Atkinson, R. & Stohlman, S. Control of central nervous system viral persistence by neutralizing antibody. J. Virol. 77, 4670–4678 (2003).
Tschen, S. I., Bergmann, C., Ramakrishna, C., Atkinson, R. & Stohlman, S. Recruitment kinetics of antibody secreting cells within the CNS following viral encephalomyelitis. J. Immunol. 168, 2922–2929 (2002). References 100 and 101 show that virus-specific ASCs are recruited and retained in the CNS after acute virus infection is controlled and, importantly, that neutralizing antibody is the crucial effector in controlling a chronic coronavirus infection of the CNS.
Tyor, W. R. & Griffin, D. E. Virus specificity and isotype expression of intraparenchymal antibody-secreting cells during Sindbis virus encephalitis in mice. J. Neuroimmunol. 48, 37–44 (1993).
Mokhtarian, F., Huan, C. M., Roman, C. & Raine, C. S. Semliki Forest virus-induced demyelination and remyelination — involvement of B cells and anti-myelin antibodies. J. Neuroimmunol. 137, 19–31 (2003).
Redwine, J. M., Buchmeier, M. J. & Evans, C. F. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Amer. J. Pathol. 159, 1219–1224 (2001).
Tyler, K. & Gonzalez-Scarano, F. Viral diseases of the central nervous system: acute infection. In Viral Pathogenesis (ed. Nathanson, N.) 837–854 (Lippincott–Rave, Philadelphia, 1997).
Gu, J. et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 202, 415–424 (2005).
Stohlman, S. A., Fleming, J. O., Brayton, P. R., Weiner, L. P. & Lai, M. M. C. Murine coronaviruses: isolation and characterization of two plaque morphology variants of the JHM neurotropic strain. J. Gen. Virol. 63, 265–275 (1982).
Navas, S. & Weiss, S. R. Murine coronavirus-induced hepatitis: JHM genetic background eliminates A59 spike-determined hepatotropism. J. Virol. 77, 4972–4978 (2003).
Rempel, J. D., Murray, S. J., Meisner, J. & Buchmeier, M. J. Mouse hepatitis virus neurovirulence: evidence of a linkage between S glycoprotein expression and immunopathology. Virology 318, 45–54 (2004).
Acknowledgements
This work was supported by National Institutes of Health grants.
Author information
Authors and Affiliations
Corresponding author
- https://www.nature.com/articles/nrmicro1343
Slider with three content items shown per slide. Use the Previous and Next buttons to navigate the slides or the slide controller buttons at the end to navigate through each slide.
- Review Article
- Published:
https://twitter.com/guty2370/status/1442821049930895363
https://articulosclaves.blogspot.com/2023/12/los-fallos-globales-causaron-millones.html
https://articulosclaves.blogspot.com/2023/12/que-esta-pasando-al-final-del-cuarto.html
https://articulosclaves.blogspot.com/2023/12/la-falta-de-coordinacion-y-medios.htmlEn este estudio hemos demostrado que los pacientes con COVID Persistente presentan una memoria celular y humoral disminuida frente al virus comparados con aquellos que no lo han desarrollado
- https://t.co/K65ZNtVSPJ
No hay comentarios:
Publicar un comentario